

Organic acidemias are among the rarer causes of dilated cardiomyopathy (DCM). They form a group of inherited metabolic diseases characterized by excessive accumulation of organic acids leading to systemic disease. Among this group, propionic acidemia is the type that frequently involves the heart. We present a case of DCM with a diagnosis of propionic acidemia. The patient gave informed consent to undergo tests and to the publication of the case, which was approved by the ethics committee of our hospital.
A 38-year-old woman was referred to the cardiology department because of a finding of DCM on echocardiography. The family history included a brother diagnosed with DCM of unidentified etiology, who died at 21 years of age while undergoing cardiac transplant (figure 1). The personal history referred to episodes of hypoglycemic ketotic crises in childhood and highlighted a diagnosis of chronic kidney disease of unclear etiology in 2015. In 2019, due to the progression of renal disease, renal transplant was performed without subsequent complications.
 and genes studied in the index case.")
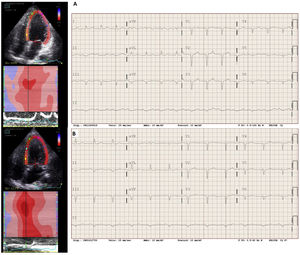
At the time of the diagnosis of DCM, the patient was in New York Heart Association (NYHA) functional class II, with a spherical dilated (end-diastolic diameter, 65mm) left ventricle (LV) with severe LV systolic dysfunction (LV ejection fraction [LVEF], 33%) and severe relaxation deficit with elevated filling pressures (E/E’, 14) (figure 2). The electrocardiogram (ECG) showed a long QTc interval (figure 2). Coronary angiography showed no lesions.
 of -9% was observed on echocardiogram and a QTc of 485ms (Bazett")
A genetic study was performed using a general DCM panel (figure 1), which was negative. Decompensated heart failure was not observed on optimized drug therapy; in March 2021, the patient was in NYHA functional class I, had achieved partial echocardiographic remission with a slightly dilated LV (end-diastolic diameter, 59mm) with a recovered LVEF (55%), and diastolic normalization (E/E’, 7), but with decreased global longitudinal strain (figure 2). Improvement was seen on electrocardiography with QT shortening (figure 2).
In July 2021, the patient was admitted to the psychiatry department for behavioral disturbances. Brain magnetic resonance imaging (MRI) showed a hypersignal in the basal ganglia, so the patient was referred to neurology. The etiological study included a urine organic acid study; we highlight increases in 3-hydroxypropionic acid (146 mmol/mol creatinine; normal, < 5) and methylcitric acid (99 mmol/mol creatinine; normal, < 5), with normal levels of methylmalonic acid. Whole exome sequencing targeting genes related to propionate and biotin metabolism disorders showed 2 heterozygous mutations in the PCCB gene (figure 1). Both are described in the Human Gene Mutation Database. As predictors of pathogenicity, and according to the criteria of the American College of Medical Genetics, they are classified as pathogenic variants, thus confirming the diagnosis of propionic acidemia.
Propionic acidemia is an inherited autosomal recessive disease characterized by the accumulation of propionate and its metabolites due to mutations in propionyl-CoA carboxylase (PCC), which is a mitochondrial enzyme that catalyzes a reaction prior to entry into the Krebs cycle. The PCC enzyme is composed of PCCA and PCCB subunits, which are encoded by the PCCA and PCCB genes. Propionic acidemia can result from mutations in these genes.
In clinical terms, the disease leads to chronic multiorgan involvement and ketotic hypoglycemia in the presence of precipitating factors (infections or fasting).1
Cardiac involvement is common in the form of cardiomyopathy and long QT syndrome. Cardiomyopathy occurs in 25% of cases,2 with a mean age at presentation of 7 years.3 It may be the only manifestation of the disease and the most frequent form of presentation is DCM (> 90%).2 The incidence of long QT syndrome varies (22%-70%)1 and increases with age. Both cardiomyopathy and a long QT interval may be caused by the toxic effects of propionate and its metabolites. The prognosis of patients with cardiomyopathy is poor, with a mortality rate of 50% due to progression of cardiomyopathy2 and sudden cardiac death due to ventricular arrhythmias. Some cases have required cardiac transplant.4
Diagnosis is based on a urine organic acid study in which elevated levels of 3-hydroxypropionate and 2-methylcitrate are observed together with normal values of methylmalonic acid.1 The diagnosis is confirmed by genetic study, detecting pathogenic variants of the PCCA or PCCB genes. Of note, these genes are not included in the recommended DCM panel.5 Although this disease is not often suspected, it causes systemic disease and particularly affects the heart: thus, some studies have suggested that these genes should be included in the general DCM panel to achieve early diagnosis.6
Propionic acidemia should be suspected in a case of DCM accompanied by a long QT interval, acute or intermittent neurological symptoms, ketotic hypoglycemia, and a family history of cardiomyopathy or sudden cardiac death.
FUNDINGD. Heine Suñer has received funding from the Instituto de Salud Carlos III through project PI18/00847 (cofinanced by the European Regional Development Fund/European Social Fund «A way to make Europe/Investing in your future»).
AUTHORS’ CONTRIBUTIONSJ Siquier wrote the original text. J Pons and A Grau reviewed the original text in detail and provided the transthoracic echocardiography images. D Heine-Suñer performed and interpreted the patient's genetic study. All authors actively participated in this case, reviewed the manuscript, and approved its submission.
CONFLICTS OF INTERESTA Grau has received honoraria from AstraZeneca for presentations at congresses, and from Novartis as an expert witness, for presentations, and attendance at congresses. M Massot-Cladera is the president of the Balearic Society of Neurology.