
Una de las causas poco habituales de miocardiopatía dilatada (MD) son las acidemias orgánicas, un grupo de enfermedades metabólicas hereditarias caracterizadas por la acumulación excesiva de ácidos orgánicos que producen una enfermedad sistémica. Dentro de este grupo, la acidemia propiónica es la que más frecuentemente produce afección cardiaca. Se presenta un caso de MD con diagnóstico de acidemia propiónica. La paciente dio el consentimiento informado para la realización de las pruebas y la publicación del caso, que fue aprobada por el comité de ética de nuestro centro.
Una mujer de 38 años fue derivada a consultas de cardiología por hallazgo de una MD en un ecocardiograma. Entre los antecedentes familiares, destaca un hermano diagnosticado de una MD de etiología no filiada, que falleció a los 21 años mientras estaba en estudio para trasplante cardiaco (figura 1). Entre los antecedentes personales, refirió episodios de crisis cetósicas hipoglucémicas en la infancia y destacó el diagnóstico de una enfermedad renal crónica de etiología no aclarada en 2015. Por progresión de la enfermedad renal, en 2019 se sometió a trasplante renal, sin complicaciones posteriores.
 y genes estudiados en el caso índice.")
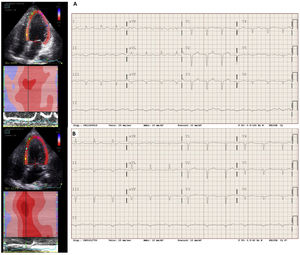
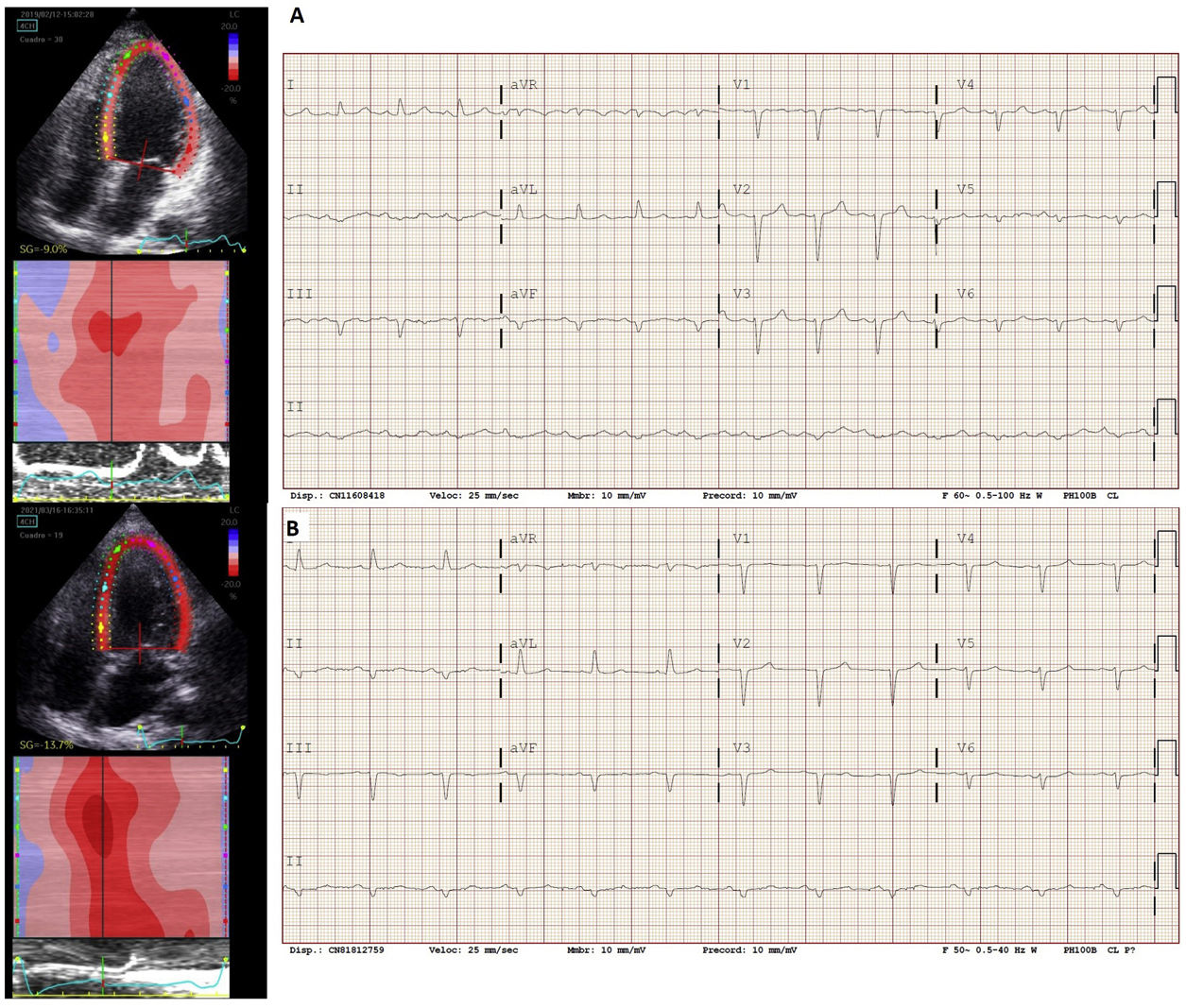
En el momento del diagnóstico de la MD estaba en clase funcional II de la New York Heart Association (NYHA), con el ventrículo izquierdo (VI) dilatado (diámetro telediastólico, 65 mm) y esférico con disfunción sistólica del VI grave (fracción de eyección del VI [FEVI], 33%) y déficit grave de relajación con presiones de llenado elevadas (E/E’, 14) (figura 2). En el ECG destacaba un intervalo QTc largo (figura 2). La coronariografía no mostró lesiones.
 de –9% y en el electrocardiograma, un QTc de 485 ms (fórmula de Bazett). B: en 2021 se observó en el ecocardiograma una mejoría del GLS (–14%) y del QTc (436 ms).")
Se realizó estudio genético con panel general de MD (figura 1), que resultó negativo. En tratamiento farmacológico optimizado, no ha tenido descompensaciones de la insuficiencia cardiaca; en marzo de 2021 se encontraba en clase funcional NYHA I, consiguió una remisión parcial ecocardiográfica, con un VI ligeramente dilatado (diámetro telediastólico, 59 mm) con una FEVI recuperada (FEVI del 55%), normalización diastólica (E/E’, 7) pero con strain longitudinal global disminuido (figura 2). Su electrocardiografía mejoró y presentaba un acortamiento del QT (figura 2).
En julio de 2021 ingresó en psiquiatría por alteración de la conducta. En la resonancia cerebral se observó una hiperseñal en los ganglios basales, por lo que se la derivó a neurología. En el estudio etiológico, se analizaron ácidos orgánicos en orina; destacaba un aumento del ácido 3-hidroxipropiónico (146mmol/mol creatinina; normal, < 5) y el ácido metilcítrico (99mmol/mol creatinina; normal, < 5), con cifras normales de ácido metilmalónico. En el estudio genético de secuenciación exómica dirigido a genes relacionados con trastornos del metabolismo del propionato y la biotina, se observaron 2 mutaciones en heterocigosis en el gen PCCB (figura 1); ambas están descritas en la Human Gene Mutation Database, y los predictores de patogenicidad, según los criterios del American College of Medical Genetics, las clasifican como variantes patogénicas, por lo que se confirmó el diagnóstico de acidemia propiónica.
La acidemia propiónica es una enfermedad hereditaria autosómica recesiva caracterizada por la acumulación de propionato y sus metabolitos debida a mutaciones en la propionil-CoA carboxilasa (PCC), una enzima mitocondrial que cataliza una reacción previa a la entrada en el ciclo de Krebs. La enzima PCC está compuesta por subunidades PCCA y PCCB, codificadas por los genes PCCA y PCCB. La acidemia propiónica puede resultar de mutaciones en estos genes.
Clínicamente produce afección crónica multiorgánica, así como crisis cetósicas hipoglucémicas ante precipitantes (infecciones o ayuno)1.
La afección cardiaca es frecuente en forma de miocardiopatía y síndrome de QT largo. La miocardiopatía se produce en un 25% de los casos2, con una media de edad a la presentación de 7 años3, puede ser la única manifestación de la enfermedad y la forma más frecuente de presentación es la MD (> 90%)2. El síndrome de QT largo tiene una incidencia variable (22-70%)1 y aumenta con la edad. Tanto la miocardiopatía como el QT largo se producen por los efectos tóxicos del propionato y sus metabolitos. El pronóstico de los pacientes con miocardiopatía es malo: la mortalidad es del 50% debido a la progresión de la miocardiopatía2, así como por muerte súbita por arritmias ventriculares. Algunos casos han requerido trasplante cardiaco4.
El diagnóstico se basa en un estudio de ácidos orgánicos en orina en el que se observarán cifras elevadas de 3-hidroxipropionato y 2-metilcitrato con valores normales de ácido metilmalónico1. El diagnóstico de confirmación es genético mediante la detección de variantes patogénicas de los genes PCCA o PCCB. Cabe destacar que dichos genes no están incluidos en el panel de MD recomendado5. Al tratarse de una enfermedad de difícil sospecha clínica, que produce una enfermedad sistémica y con un comportamiento agresivo en el corazón, algunos estudios indican que estos genes deberían incluirse en el panel general de MD para lograr un diagnóstico precoz6.
Se debe sospechar una acidemia propiónica ante un caso de MD que se acompañe de QT largo, síntomas neurológicos agudos o intermitentes, crisis cetósicas hipoglucémicas y antecedentes familiares de miocardiopatía o muerte súbita.
FINANCIACIÓND. Heine Suñer ha recibido financiación del Instituto de Salud Carlos III a través del proyecto PI18/00847 (cofinanciado por el Fondo Europeo de Desarrollo Regional/Fondo Social Europeo Una manera de hacer Europa/Invirtiendo en tu futuro).
CONTRIBUCIÓN DE LOS AUTORESJ. Siquier redactó el primer texto original. J. Pons y A. Grau revisaron detalladamente el texto original y aportaron las imágenes de ecocardiografía transtorácica. D. Heine-Suñer realizó e interpretó el estudio genético de la paciente. Todos los autores participaron activamente en este caso, revisaron el manuscrito y aprobaron su presentación.
CONFLICTO DE INTERESESA. Grau ha recibido honorarios de AstraZeneca por ponencias en congresos, y de Novartis como testigo experto, por ponencias y asistencia a congresos. M. Massot-Cladera es la presidenta de la Sociedad Balear de Neurología.