To the Editor,
Pompe disease, also known as glycogen storage disease type II, is caused by glycogen accumulation due to a deficiency of the lysosomal acid alpha-glucosidase enzyme by which it is degraded.1 A total or partial deficiency of this enzyme causes lysosomal glycogen storage leading to a systemic disorder characterized by cardiomyopathy, muscle weakness, hypotonia, and respiratory disorders.1, 2 Three forms of presentation have been described according to the age at which clinical signs appear: in adulthood, adolescence, or infancy. The latter, characterized by very severe or even total enzyme deficiency, usually manifests in the first months of life and if left untreated causes death within 1 year.1, 2 This form is specifically characterized by severe cardiomyopathy as a result of glycogen accumulation and manifests as hypertrophic cardiomyopathy on electrocardiography and echocardiography.2 The analysis of enzymatic activity in cultured skin fibroblasts is the diagnostic procedure with the greatest sensitivity and specificity, although it is complex and can delay diagnosis between 4 weeks and 6 weeks.1 Treatment with recombinant human alpha-glucosidase (rhGAA) is effective regardless of patient age at onset. This approach has been tried even in infants younger than 3 months, obtaining good results in terms of reducing the severity of cardiomyopathy and prolonging survival.1, 3 Regarding cardiomyopathy, various studies have shown that rhGAA treatment leads to improvements in electrocardiographic abnormalities,4, 5 reductions in ventricular mass, and improvements in ventricular function.3, 4, 6 Furthermore, the first effects can be detected within the first weeks of treatment.
If prognosis, procedural delays in assessing enzymatic activity, and the advantages of early treatment are taken into account, it is pertinent to ask whether enzyme replacement therapy could be initiated in neonates with a high suspicion of Pompe disease prior to diagnostic confirmation.
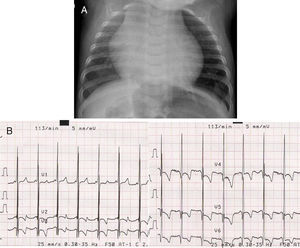
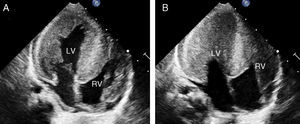
We present the case of a neonate (3 months) who was admitted with symptoms of respiratory failure and presenting feeding intolerance and tachypnea. Hypotonia and generalized areflexia were observed during examination. Blood analysis showed elevated creatine kinase levels. Chest x-ray showed massive cardiomegaly (Figure 1A) and electrocardiogram showed a pattern of left ventricular hypertrophy and a short PR interval (Figure 1B). Echocardiography showed very severe generalized left ventricular hypertrophy (interventricular septum, 56mm/m2) that also affected the right ventricle (Figure 2A) and caused the disappearance of the ventricular cavity in systole (Figure 2B). Based on this, Pompe disease was suspected and a skin sample was taken to analyze enzyme deficiency in cultured skin fibroblasts.

Figure 1. A, Chest x-ray. B, Electrocardiogram (performed at 5mm/mV): Signs of left ventricular hypertrophy and a short PR interval in precordial leads. These abnormalities are characteristic of cardiomyopathy associated with Pompe disease.

Figure 2. Transthoracic echocardiogram, apical four-chamber view. LV, left ventricle; RV, right ventricle.
While waiting for the test results, which confirmed a residual enzymatic activity of less than 1%, the patient died of cardiorespiratory failure.
Would high clinical suspicion of the disease have been sufficient to initiate enzyme replacement therapy? In cases such as the one presented in which the clinical manifestations and complementary tests strengthen the suspicion of Pompe disease, and the clinical course suggests an imminent fatal outcome, we suggest that the empirical administration of rhGAA treatment could be of value while awaiting diagnostic confirmation. This approach should be considered if we also take into account that, in its infantile form, the low incidence of this disease confers high specificity and positive predictive value when clinical suspicion is high. Based on these premises, the possible adverse effects that can occur after rhGAA administration become an acceptable risk. Although some of these effects are severe they have low incidence,1 which is of particular interest especially in cases where the suspected diagnosis is not confirmed by the test results.
.
Corresponding author: jnlsbnll@hotmail.com