Primary hyperoxaluria (PH) is an infrequent condition, with autosomal recessive inheritance, and consisting of abnormal oxalate metabolism with calcium depositions in the kidneys that lead to a decline in glomerular filtration rate (GFR) and progressive renal insufficiency (RI).1,2 This metabolic defect takes different forms. Type PH (PH1) is produced by an enzyme abnormality in the liver and is frequently accompanied by heart disorders caused by oxalate depositions in the cardiac tissue.2 Dilated cardiomyopathy has been described, with various degrees of ventricular dysfunction, stroke—due to embolization of myocardial depositions—and conduction-specific tissue abnormalities, which appear as conduction abnormalities and potentially fatal arrhythmias.2 Treatment of PH1 is by liver transplantation to recover the deficient enzyme activity.
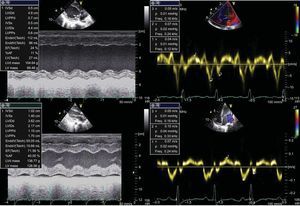
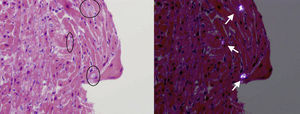
A 3-year-old girl (weight, 10.4kg) of Pakistani origin, admitted for an RI study (urea, 543 mg/dL; creatinine, 11.6 mg/dL; GFR, 14.3%), was diagnosed with PH (plasma oxalate, 12.6 mg/L; urinary oxalic acid, 540.4 mmol/mol). The genetic study detected homozygosis due to a previously undescribed mutation consisting of a change in codon 164 from glutamine to arginine (p.Q164R), which codifies a nonfunctioning protein demonstrated in vitro. These findings are diagnostic of PH1. Clinically, our patient had episodes of acute lung edema related to intravenous fluid administration. Echocardiography revealed left ventricular dilatation (z-score, +4) with severe ventricular dysfunction (left ventricular ejection fraction [LVEF], 23%) (Fig. 1). Suspected dilated cardiomyopathy secondary to PH1 led to a myocardial biopsy, which showed the presence of crystals with radial striations, visible in polarized light, associated with heart disease caused by PH (Fig. 2).
 and tissue Doppler (right). The lower images show recovery of ventricular function at 18 months after combined liver and kidney transplantation.")
. Right: the same image with polarized light to improve visibility of the depositions (arrows).")
Given the high surgical risk of combined liver and kidney transplantation in the presence of severe ventricular dysfunction, a myocardial reserve study was performed using dobutamine stress echocardiography and following the American Heart Association protocol.3 This showed improved left ventricular function up to 55%-60% LVEF with dobutamine at 30μg/kg/min but the test had to be interrupted due to a hypertensive crisis with transient neurologic symptoms.
On the basis of these results and the current literature, the patient was listed for combined liver and kidney transplantation, which took place 1 month later.
In the immediate postoperative period, left ventricular dilatation persisted with dysfunction (25%-30% LVEF) but subsequent follow-up showed increased myocardial thickness and progressive LVEF recovery; at 6 and 18 months post-transplantation LVEF was 68% and 71.6% respectively, with ventricular diameters normal for the patient's age (Fig. 1).
PH1 is caused by a chromosome 2 abnormality, associated with q36-37, leading to absent, decreased or dysfunctional glycolate aminotransferase enzyme hepatic activity. This abnormality leads to excessive synthesis of oxalate and glycolate, which are deposited in the kidneys as calcium salts producing a progressive decline in GFR and, finally, terminal RI.1,4 As GFR declines, oxalate concentration increases and “systemic oxalosis” follows, leading to accumulated oxalate depositions in organs such as the pulmonary vasculature, cardiac tissue, retina or osteoarticular system.2 It has been clearly established that a <20-40mL/min/m2 GFR leads to increased oxalate and favors oxalosis.4
The presentation and clinical course of PH1 vary substantially within a single genetic mutation,4 hampering prediction of the clinical course. Prognosis worsens in the presence of multisystemic conditions, especially cardiac or pulmonary involvement.2 Children are the subgroup at highest risk of rapidly progressive disease, showing RI and oxalosis when very young.5
Initially kidney transplantation was the only therapeutic option indicated but the recurrence rate was high.1,4,5 Later, liver or combined liver and kidney transplantation (in patients with RI) was found to give better long-term results (overall survival and graft survival4), attributable to correction of the enzymatic defect.4–6 Combining hemodialysis and peritoneal dialysis prior to transplantation facilitates the mobilization of oxalic depositions with decreased oxalate and favors better short- and long-term kidney graft survival.5 No consensus exists on the GFR values that establish the indication for liver-only transplantation but some groups4,5 report excellent results with >50mL/min/m2 GFR.
Few instances of patients with dilated cardiomyopathy secondary to PH1 and undergoing combined liver and kidney transplantation have been described, although most show LVEF recovery following transplantation.
The ideal therapeutic option would be to perform liver transplantation before RI. However, when RI is established, combined liver and kidney transplantation is needed. In patients with severe cardiac disease, combined heart-liver-kidney transplantation could be considered. However, the frequent reversibility described even in extreme cases indicates combined liver and kidney transplantation. Although not previously described, in our patient, stress echocardiography made an important contribution to decision-making as it showed myocardial reserve prior to transplantation.
.