
Pheochromocytoma and paraganglioma are neuroendocrine tumors (NETs) that produce catecholamines derived from neural crest cells, localized in the adrenal medulla (90%) or in extra-adrenal chromaffin tissue (10%). Their prevalence is between 0.2% and 0.6% in hypertensive adults, 5% in adrenal incidentalomas, and 0.05% to 0.1% in autopsy series.1 Although they usually present as isolated tumors, they can be associated with hereditary syndromes such as multiple endocrine neoplasms, neurofibromatosis, or von Hippel Lindau syndrome. Furthermore, there have been case reports and small series highlighting the association between cyanotic congenital heart defects (CCHD) and NETs.2 Recently, Opotowsky et al.3 reported an increased risk of NETs in patients with CCHD in a multicenter study in which the role of chronic hypoxia in association with genetic susceptibility was proposed as the underlying pathogenic mechanism for these tumors.
A retrospective analysis was performed of 3311 adults with congenital heart defects, 173 with CCHD, and 33 with Eisenmenger syndrome in a national referral center for adult congenital heart defects. The median length of follow-up was 25 years (range, 10.5 years). All patients with suspected NET under follow-up in the endocrinology department underwent computed tomography and metaiodobenzylguanidine scintigraphy, with monitoring of catecholamines in urine.
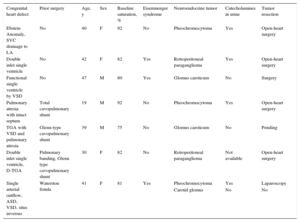
A total of 8 NETs were identified in 7 patients with CCHD (4.6%) (Table 1). Overall, 48.8% were men and the median age was 40.0 years (range, 19.0-47.0 years). All patients had active cyanosis at the time of diagnosis (mean, 36.0 ± 11.3 years), including 1 patient with Fontan circulation with venovenous collaterals. The mean baseline arterial oxygen saturation was 83.4% ± 6.3%, mean hemoglobin was 18.1 ± 2.0g/dL, and mean hematocrit was 66.5% ± 7.3%. Three patients had Eisenmenger syndrome (9.1% of all patients had this syndrome). On analysis of all patients with congenital heart defects, including those with noncyanotic disease, 1 additional NET was identified in a patient with partial anomalous pulmonary venous drainage (0.2% overall).
Characteristics of Patients With Congenital Heart Defects and Neuroendocrine Tumors
| Congenital heart defect | Prior surgery | Age, y | Sex | Baseline saturation, % | Eisenmenger syndrome | Neuroendocrine tumor | Catecholamines in urine | Tumor resection |
|---|---|---|---|---|---|---|---|---|
| Ebstein Anomaly, SVC drainage to LA | No | 40 | F | 92 | No | Pheochromocytoma | Yes | Open-heart surgery |
| Double inlet single ventricle | No | 42 | F | 82 | Yes | Retroperitoneal paraganglioma | Yes | Open-heart surgery |
| Functional single ventricle by VSD | No | 47 | M | 80 | Yes | Glomus caroticum | No | Surgery |
| Pulmonary atresia with intact septum | Total cavopulmonary shunt | 19 | M | 92 | No | Pheochromocytoma | Yes | Open-heart surgery |
| TGA with VSD and pulmonary atresia | Glenn-type cavopulmonary shunt | 39 | M | 75 | No | Glomus caroticum | No | Pending |
| Double inlet single ventricle, D-TGA | Pulmonary banding, Glenn type cavopulmonary shunt | 30 | F | 82 | No | Retroperitoneal paraganglioma | Not available | Open-heart surgery |
| Single arterial outflow, ASD, VSD, situs inversus | Waterston fistula | 41 | F | 81 | Yes | Pheochromocytoma | Yes | Laparoscopy |
| Carotid glomus | No | No |
ASD, atrial septal defect; F, female; LA, left atrium; M, male; SVC, superior vena cava; TGA, transposition of the great arteries; VSD, ventricular septal defect.
Among patients with CCHD, 3 cases of pheochromocytoma (2 in the left adrenal gland and 1 in the right adrenal gland) and 5 cases of paraganglioma were diagnosed (3 cases of glomus caroticum [GC] and 2 cases of extra-adrenal paraganglioma in the retroperitoneal space). One patient (14.3%) had a pheochromocytoma and as well as GC. No other multisystemic syndromes associated with NET were detected. The form of presentation was hypertension in 3 patients with pheochromocytoma, abdominal pain in 1 patient with retroperitoneal paraganglioma, and hypoacusia in 1 patient with GC; diagnosis was coincidental in 2 patients. Catecholamines were detected in urine in all patients with pheochromocytoma and in the 1 patient with retroperitoneal paraganglioma, with elevated metanephrine in 3 patients (50%), elevated normetanephrine in 3 patients (50%), and elevated vanilmandelic acid in 2 patients (33.3%) (Table 2). Three patients (60%) had simultaneous elevation of at least 2 metabolites in urine. Data were not available for 1 patient with retroperitoneal paraganglioma because the diagnosis was made at a different hospital.
Detection of Catecholamines in Urine
| Congenital heart defect | Tumor | Metanephrine | Normetanephrine | Vanilmandelic acid |
|---|---|---|---|---|
| Ebstein Anomaly, SVC drainage to LA | Pheochromocytoma | + | + | – |
| Double inlet single ventricle | Retroperitoneal paraganglioma | – | + | – |
| Functional single ventricle by VSD | Glomus caroticum | – | – | – |
| Pulmonary atresia with intact septum | Pheochromocytoma | + | – | + |
| TGA with VSD and pulmonary atresia | Glomus caroticum | – | – | – |
| Double inlet single ventricle, D-TGA | Retroperitoneal paraganglioma | Not available | Not available | Not available |
| Single arterial outflow, ASD, VSD, situs inversus | Pheochromocytoma, glomus caroticum | + | + | + |
ASD, atrial septal defect; LA, left atrium; SVC, superior vena cava; TGA, transposition of the great arteries; VSD, ventricular septal defect.
After confirmation of the diagnosis, the tumor was resected by urologists, ear-nose-throat specialists, or general surgeons after treatment with alpha blockers and infusion of saline solution. In the patient with 2 tumors, only the pheochromocytoma was resected as a conservative approach was chosen with the GC. Postoperative complications included acute pulmonary edema in a patient with pheochromocytoma who had fluid overload and transient ischemic attack, without sequelae, in another patient with GC. No patients died in hospital or during follow-up. In the histopathological analysis, only 1 sample showed findings suggestive of malignancy. None of the patients showed metastasis.
Here, we report our experience with NET in patients with CCHD in the largest Spanish series published to our knowledge. An increased prevalence of NET was observed, especially in patients with Eisenmenger syndrome, probably due to chronic exposure to hypoxia, which stimulates formation of erythropoietin and growth factors regulated by hypoxia inducible factor 1 (vascular endothelial growth factor and platelet-derived growth factor), thus favoring tumor genesis.4 Our patients included those with uncorrected congenital heart defects, palliative corrections, and venovenous collateral flow after a Fontan procedure. All had active cyanosis at the time of diagnosis, defined as clinical cyanosis, and all had low baseline arterial oxygen saturation (in most patients<83%). In addition to hypoxia, genetic susceptibility has also been reported as a triggering factor, although in our series, only 1 patient had multiple NETs and the incidence (14.3%) was significantly lower than that published in previous series (up to 39%).3
The onset of NET can lead to general and hemodynamic deterioration in these patients and therefore early diagnosis and treatment are essential to reduce the risk of complications. The signs and symptoms of these tumors overlap with those associated with complications of CCHD, such as arrhythmias, hypertension, and heart failure. Thus, the presence of NET should be suspected with the onset of new symptoms in patients with CCHD, even after surgical correction, as these tumors are a potentially treatable cause of clinical deterioration in these patients. A multidisciplinary approach, with tumor resection in a specialized center, is associated with a high success rate, even in this population at risk.2 This treatment is effective and is associated with good short- and long-term prognosis.
CONFLICTS OF INTERESTÁ. Sánchez-Recalde is an Associate Editor of Revista Española de Cardiología.