Generalized arterial calcification of infancy (GACI) (Online Mendelian Inheritance in Man [OMIM] 208000) is a rare disorder affecting 1 in every 391000 to 566000 newborns. The condition is characterized by abnormal tissue mineralization, producing calcium build-up in the internal elastic lamina of medium-sized and large arteries of the body and proliferation in the tunica intima of muscular arteries, leading to narrowing of the arterial lumen and clinical repercussions in the territories perfused by those arteries.1
Clinical symptoms are myocardial ischemia, as well as vascular and periarticular calcifications in soft tissues. The diagnosis is confirmed by genetic study. In 70% of published cases, a mutation has been identified in the ENPP1 gene (OMIM 173335). This gene encodes ectonucleotide pyrophosphate/phosphodiesterase 1, which produces inorganic pyrophosphate, an essential physiologic inhibitor of arterial calcification. In the rest of cases, mutations have been identified in the ABCC6 gene, which encodes the MRP6 protein, a transmembrane adenosine triphosphate-binding cassette (ABC) protein transporter.2
There is no specific treatment, although bisphosphonates appear to increase survival in some patients. The prognosis depends on the extent of the calcification and associated complications, which lead to early death in many of these patients.3
Because only a few clinical cases have been published with a genetic study, we consider this report to be of interest.
We describe the case of an infant born at 33 weeks of gestation and hospitalized in the neonatal unit due to prematurity.
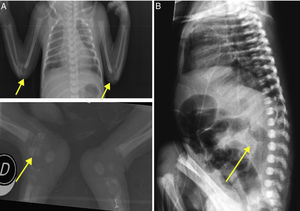
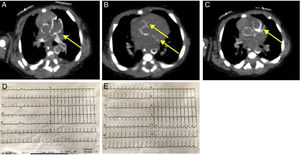
The initial examination revealed considerable limitation for elbow extension and hip mobilization. Limb X-rays disclosed periarticular calcifications (figure 1A). An electrocardiogram was also performed (figure 2D). Cardiac ultrasound on the third day of life showed a normal structure with preserved myocardial function, but hyperechogenicity in the coronary arteries. On chest and abdominal computed tomography (CT), calcification was observed from the ascending aorta to the iliac branches and carotid and humeral arteries, with coronary and lung damage also visualized (figure 2A-C). Calcium and phosphate metabolism was normal.
. B, abdominal X-ray: calcification of the descending aorta.")
 and circumflex (bottom) arteries. C, left anterior descending artery. D, initial electrocardiogram. E, subsequent electrocardiogram (ST-segment depression in V5 and V6 indicated by arrows).")
Computed tomography scan and electrocardiogram. A, calcification of the pulmonary artery and ascending aorta. B, right coronary (top) and circumflex (bottom) arteries. C, left anterior descending artery. D, initial electrocardiogram. E, subsequent electrocardiogram (ST-segment depression in V5 and V6 indicated by arrows).
On day 14 of life, the patient experienced a sudden episode of clinical worsening with respiratory pauses and evidence of low cardiac output, generalized pallor, slow filling, severe mixed acidosis, and associated severe hyperlactacidemia. Repeat echocardiography showed severe ventricular dysfunction with a left ventricular ejection fraction of 27%, as well as higher hypocontractility of the inferolateral segments. Electrocardiography revealed ST-segment depression in leads V5 and V6 (figure 2E). Cardiac biomarkers were elevated. Therefore, the diagnosis was non–ST-segment elevation acute myocardial infarction, complicated by cardiogenic shock. Inotropic support was started, and the patient was transferred to a tertiary hospital.
Following admission, ventricular function was partially restored and, in view of the probable GACI diagnosis, intravenous pamidronate therapy was started. The patient experienced progressive clinical worsening and died at 34 days of life.
In our patient, the genetic study was based on next-generation sequencing of coding exons and flanking intronic regions (NextGeneDx/Imegen panel, Spain, for GACI) of the ABCC6 and ENPP1 genes, using the Nextera XT kit (Illumina, United States). The study also included sequencing (MiSeq System, Illumina, United States) of libraries (2×150) plus Sanger sequencing of regions of interest with coverage of <100×(exons 1 and 8 of ENPP1 and exon 2 of ABCC6). The study revealed common variants and a heterozygous variant previously described4 in the ENPP1 gene, at the end of exon 4, c.556G>C, p.Gly186Arg, which is considered pathogenic (Sorting Intolerant from Tolerant Algorithm [SIFT], Polyphen2-HDIV [HumDiv; 0.999], Polyphen2-HVAR [HumVar; 0.986], MutationTaster [disease causing] and Combined Annotation Dependent Depletion [CADD] score of 34) due to a highly intact domain of somatomedin B2 protein. Another heterozygous mutation was found in intron 13 (c.1405+5G>C), not listed in the Genome Aggregation Database (gnomAD) and not previously described in the literature. These variants segregated in both parents, who were healthy, and each of these variants was found in an allele from each parent. The patient developed the disease due to compound heterozygosity (with trans configuration) similar to autosomal recessive inheritance, already described in several families affected by GACI.2,5 However, according to in silico prediction (Human Splicing Finder) for this new variant, the variant would produce rupture of the splice donor site (variation, –40.36%), thus potentially affecting proper processing of the messenger RNA and favoring the pathogenic effect of this change. Nevertheless, some patients have only 1 pathogenic mutation of ENPP1 in a single allele and a consistent pathologic phenotype.2
GACI is a very rare disorder, with variable clinical symptoms depending on the extent and location of the territory affected by vascular calcification.
Mutations in the ENPP1 and ABCC6 genes have been described as the cause in the case reports published to date.2,3
The prognosis is fatal in most patients, and clinical suspicion is important to direct a specific genetic study, as the family can receive appropriate genetic counseling once the mutation has been identified.5
We would like to thank the family for allowing us to publish this case report.