
Phenotypic overlapping of congenital heart disease, noncompaction cardiomyopathy, and arrhythmogenic cardiomyopathy is uncommon. In Revista Española de Cardiología, Bermúdez-Jiménez et al.1 published an article describing a family with a mutation in the NKX2.5 gene (p.Glu167Lys) showing this phenotype. We consider it appropriate to further highlight the risk of sudden death associated with NKX2.5 mutations related to this phenotype. To this end, we describe a family attended in our center
The NKX2.5 gene codes for a transcription factor containing 3 domains that are implicated in cardiac development.2 The homeobox domain is needed for interactions with DNA and other transcription factors. NKX2.5 mutations have been associated with cardiac septal defects, conduction defects, and noncompaction cardiomyopathy.
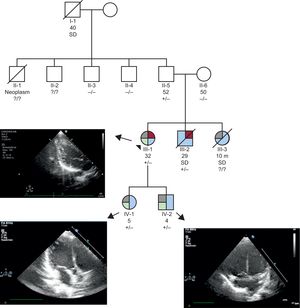
We present the case of a 30-year-old woman, whose paternal grandfather had died suddenly in his sleep at the age of 40 years. She was referred to our center for study after her brother experienced cardiac arrest. Her sister had died suddenly in childhood in the postoperative period following atrial septal defect (ASD) repair. Her brother experienced cardiac arrest in his sleep, with asystole followed by ventricular fibrillation after resuscitation maneuvers. Echocardiography detected mild left ventricular dilation, with evidence of noncompaction, moderate-severe systolic dysfunction, and an atrial septal aneurysm with no shunting. Electrocardiography findings were normal. He ultimately died due to hypoxic-ischemic encephalopathy. Autopsy was not performed. His father and mother showed no abnormalities on echocardiography or 24-hour Holter monitoring.
Our patient had undergone surgical repair of an ostium secundum ASD in infancy. Cardiac magnetic resonance showed left ventricular noncompaction, with a left ventricular ejection fraction at the lower limit of normal. First-degree atrioventricular block was observed, with no other abnormalities on Holter monitoring.
The patient is the mother of 2 children (5 and 4 years of age) diagnosed with ostium secundum ASD and hypertrabeculation on echocardiography, with no evidence of noncompaction and a prolonged PR interval for their age. The defects have not been treated to date.
Genetic study of her deceased brother was performed, using a new-generation sequencing panel. Exons from 268 genes were included, and the analysis was focused on 16 of them: ACTC1, CASQ2, DMD, DTNA, HCN4, LDB3, LMNA, MYBPC3, MYH7, NKX2-5, SCN5A, TAZ, TNNI3, TNNT2, TPM1 and VCL. A variant in heterozygosity was identified in NKX2.5 (p.Lys183Asn). The mutation has not been described previously in public databases of the general population nor has it been indicated as deleterious by bioinformatic predictors. The p.Lys183Asn variant affects a residue with high evolutionary conservation (Figure 1). Our patient, her children, and her father are all carriers of the mutation, whereas her mother is not a carrier. Cardiac magnetic resonance examination of the father disclosed no relevant findings. Two uncles were also studied, with negative results. The single positive finding (ECG, imaging) was apical hypertrabeculation in 1 of them (Figure 2).


Cosegregation of the mutation with the phenotype in 4 affected individuals in the family, with variable expressivity and penetrance, provides evidence of its probable pathogenicity. The finding of a carrier without the phenotype is consistent with the incomplete penetrance seen in other NKX2.5 mutations. In the differential diagnosis of sudden death in the patient's grandfather (subject I-1), it would be pertinent to include other causes, such as ischemic heart disease.
p.Lys183Asn is located in the NKX2.5 homeobox (amino acids 138-197). A few variants in this region have been associated with the development of ASD and conduction defects, with variable penetrance and expressivity, and most show evidence of cosegregation (eg, p.Leu171Pro3, p.Arg190His3).
Until the description of p.Glu167Lys,1 the single NKX2.5 mutation associated with a phenotype comprised of noncompaction, ASD, conduction defects, and sudden death was found in the homeobox domain of the protein (p.Leu171Argfs*7). This variant, with variable penetrance and expressivity, was described by Ouyang et al.4 in a family from the United States. One of the carriers experienced sudden death.
In a European Society of Cardiology congress, Maury et al.5 presented a series of 48 patients (18 families), carriers of NKX2.5 mutations and with 8 sudden deaths. Among the carriers, 75% had ASD and 90% had conduction defects.5 Five carriers met the criteria for noncompaction cardiomyopathy.
Our patient underwent implantation of a single-chamber defibrillator for primary prevention. The device recorded recurrent episodes of nonsustained polymorphic ventricular tachycardia and 2 appropriate shocks following degeneration to ventricular fibrillation. Treatment with sotalol was started, and there have been no recurrences in the last 7 months.
The findings in this family, together with those from the family of Bermúdez-Jiménez et al.,1 suggest that mutations in this region of NKX2.5 (homeobox) can lead to ASD, conduction defects, and ventricular noncompaction, and are associated with a high risk of sudden death. In contrast to the family reported by these authors, which showed features consistent with arrhythmogenic cardiomyopathy (carriers of a DSP mutation), there was no second mutational event in our patient that could be related to the risk of sudden death and make this association more plausible.
Detection of noncompaction in patients with ASD could alert to the possible presence of this entity, which has a malignant character.