Palabras clave
INTRODUCCIÓN
De manera muy simplificada y reduccionista, la aterosclerosis puede definirse como el resultado del desequilibro entre la entrada y la salida de colesterol en la pared arterial, con un predominio de la primera. Como puede verse en la figura 1, el principal implicado en la entrada de colesterol en la pared arterial es la lipoproteína de baja densidad (LDL), mientras que la principal encargada de la salida del colesterol del vaso arterial es la lipoproteína de alta densidad (HDL). Tanto las concentraciones sistémicas (torrente circulatorio) altas de colesterol de las LDL (cLDL) como las bajas de colesterol de las HDL (cHDL) se han asociado de forma constante con el desarrollo de aterosclerosis. Tras este descubrimiento, se han probado múltiples terapias que intentan prevenir el desarrollo de la enfermedad mediante una reducción de las LDL o un incremento de las HDL. Si bien la reducción de LDL, fundamentalmente mediante estatinas, se ha establecido como terapia estándar para la prevención primaria y secundaria de eventos aterotrombóticos, del incremento de las HDL aún no se ha demostrado de forma contundente un efecto beneficioso. En este artículo se revisa y se pone en perspectiva histórica la biología de las HDL, las intervenciones disponibles para incrementarlas y su potencial efecto beneficioso.

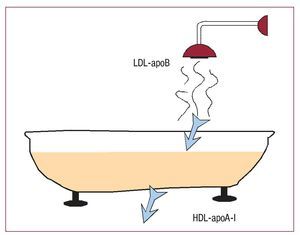
Fig. 1. Esquema representativo de los principales implicados en la entrada y la salida de colesterol. La bañera representa la pared del vaso. La principal encargada de la entrada de colesterol en el vaso es la lipoproteína de baja densidad (LDL), guiada por su principal proteína, la apolipoproteína (apo) B. La lipoproteína de alta densidad (HDL), por el contrario, es la principal encargada de la extracción de colesterol de la pared arterial mediante la interacción de su principal proteína, la apoA-I, con receptores presentes en el macrófago.
FISIOPATOLOGÍA DE LA ATEROSCLEROSIS
La relación entre una dieta rica en grasas (colesterol) y la afección aterosclerótica fue aceptada hace muchos años. Sin embargo, no fue hasta principios del siglo XX cuando Ignatowski lo demostró científicamente por primera vez1. Ese autor fue capaz de generar lesiones vasculares ateroscleróticas en conejos mediante la administración de una dieta rica en leche y yema de huevo. Otro científico ruso, Nikolai Anitschkow, identificó al colesterol como el componente de la dieta que causa el desarrollo de las lesiones ateroscleróticas2.
La enfermedad aterotrombótica es la principal causa de mortalidad en países occidentales y está convirtiéndose en la primera en países en desarrollo. Por ello, la investigación en el campo de la aterosclerosis es fundamental para poder atajar esta epidemia mundial.
La aterosclerosis es una enfermedad sistémica con manifestaciones clínicas locales3 y se caracteriza principalmente por un depósito de lípidos en la pared de las arterias de mediano y gran calibre. Las altas concentraciones de cLDL circulante inducen una acumulación de éste en la íntima de las arterias. Estas pequeñas partículas de LDL se depositan en las zonas intimales ricas en proteoglucanos y convergen en agregados de cLDL. La unión de las partículas de cLDL a los proteoglucanos promueve un «secuestro» del cLDL, lo que prolonga el contacto del cLDL con la íntima arterial4. La unión de las partículas de cLDL a los proteoglucanos hace que las primeras sean más susceptibles a la oxidación. La oxidación del cLDL se considera un evento esencial en la patogenia de la aterosclerosis5. La unión prolongada de cLDL (oxidado) a la capa íntima arterial, así como la exposición a diferentes factores de riesgo (hipertensión arterial, tabaquismo, diabetes mellitus, etc.), puede inducir un mal funcionamiento del endotelio (disfunción endotelial)6, que es el proceso patológico más precoz de la aterosclerosis7. Una vez dañado el endotelio, se produce una infiltración de cLDL en las capas arteriales más profundas. En un intento de atajar esta «invasión» del cLDL, los monocitos/ macrófagos circulantes penetran en la pared arterial, fagocitan el cLDL oxidado y se transforman en células cargadas de colesterol. Cuando los macrófagos están repletos de colesterol, se convierten en células espumosas. Debido a que el colesterol no es fácilmente metabolizable fuera del hígado, la continua acumulación de éste dentro de las células hace que, ante su fracaso vital, entren en apoptosis, con la consiguiente liberación de sustancias protrombóticas, como el factor tisular (TF)8. Tras la muerte de las células espumosas-macrófagos, el colesterol se libera de nuevo a la pared arterial y forma el núcleo lipídico de la placa, con lo que se perpetúa el proceso9. Además, el colesterol puede cristalizar, factor que se ha identificado recientemente como desestabilizador de la placa10.
Aparte de la acumulación lipídica, el reclutamiento de células inflamatorias adicionales a monocitos/macrófagos es un factor importante en la aterogénesis. De este modo, la celularidad de las lesiones ateroscleróticas se va modificando según va avanzando la enfermedad. Las lesiones iniciales (tipo fatty streaks) son lesiones en las que casi exclusivamente hay macrófagos cargados de colesterol y alguna célula leucocitaria. Según avanza la enfermedad, la lesión se hace más compleja, con diferentes tipos celulares y mucho material extracelular. A medida que el núcleo lipídico de la placa de ateroma crece por acumulación de partículas de cLDL y macrófagos, células de músculo liso migran desde la media hacia la íntima. Estas células producen y segregan colágeno y elementos fibrosos de la matriz extracelular, con lo que se forma la envoltura fibrosa de las placas fibroateromatosas.
Inicialmente estas placas no disminuyen el lumen vascular, porque hay un agrandamiento compensatorio de la pared vascular (remodelado positivo)11. La placa crece de forma excéntrica y se produce el adelgazamiento de la media y la adventicia, hasta que el agrandamiento compensatorio ya no puede proseguir, momento en el cual empieza a crecer hacia el interior de la luz vascular y deteriora el flujo sanguíneo.
La mayoría de los eventos cardiovasculares agudos no los causa el estrechamiento progresivo del lumen vascular, sino las complicaciones en la placa aterosclerótica (rotura, ulceración, hemorragia, erosión) que producen la oclusión vascular aguda por trombosis del vaso. Las placas con alto contenido lipídico también se han denominado «inestables», «vulnerables» o «de alto riesgo», por su propensión a romperse y facilitar un evento agudo. La manifestación clínica aterosclerótica se caracteriza fundamentalmente por la formación de un trombo agudo (oclusivo) anclado en la placa rota o erosionada, por ello el término aterotrombosis define de forma más integral la naturaleza de la enfermedad.
ATEROTROMBOSIS: IMPORTANCIA DE LA COMPOSICIÓN DE LA PLACA
«El hombre vive con la aterosclerosis, pero muere de trombosis.»
Si bien la acumulación de lípidos en las paredes arteriales es el marco de la aterosclerosis, la mera presencia de placas de ateroma no genera per se síntoma alguno. Las manifestaciones clínicas de la enfermedad son secundarias a roturas de placas de ateroma con superimposición de un trombo. En función de la limitación del trombo al flujo arterial, las manifestaciones serán de mayor o menor gravedad. La trombosis es el mecanismo fundamental de transición de un estado crónico-latente de la enfermedad a un estado agudo sintomático.
Las placas consideradas clásicamente como de alto riesgo son las que están compuestas generalmente por un cuerpo lipídico abundante, separado de la luz del vaso por una fina capa de matriz extracelular cubierta por el endotelio12. La rotura de las placas de ateroma parece estar dictaminada tanto por unos mecanismos pasivos (fuerzas físicas de cizallamiento) como activos (infiltración y activación de macrófagos, liberación de enzimas proteolíticas, infiltración de células inflamatorias, etc.). Especial interés tiene la presencia de las metaloproteinasas de la matriz (MMP). Las MMP son enzimas que degradan la matriz extracelular de la placa y hacen que ésta sea más inestable y, por lo tanto, más propensa a romperse. Se ha observado que las MMP se expresan en las lesiones ateroscleróticas precisamente en las regiones con mayor propensión a romperse, donde se localizan junto con macrófagos, lo que confirma su participación en la inestabiliad de la placa13.
Los mecanismos que modulan la trombosis, tanto locales como sistémicos, son muy importantes en esta enfermedad. Los factores externos a la placa (factores sistémicos) también modulan la vulnerabilidad de la placa, así como su trombogenicidad una vez rota. De hecho, algunos factores sistémicos, como elevadas concentraciones de cLDL, hábito tabáquico, hiperglucemia, diabetes y otros, se asocian con un aumento de la coagulabilidad de la sangre14. Algunos estudios recientes han demostrado que las células endoteliales que recubren las placas ateroscleróticas pueden tornarse procoagulantes a raíz de una inducción apoptótica15.
En las últimas décadas se ha puesto de manifiesto la importancia de la inflamación en la aterotrombosis. De hecho la presencia, tanto local como sistémica, de marcadores de inflamación se correlaciona con la vulnerabilidad del paciente (riesgo de sufrir un evento aterotrombótico)16.
Por todo lo aquí comentado, queda patente que la vulnerabilidad de las placas de ateroma está modelada por factores tanto locales como sistémicos. Se puede conseguir reducción tanto local como sistémica de la vulnerabilidad de las placas (estabilización). La estabilización de las placas de ateroma es un objetivo que se puede conseguir mediante fármacos. Las estatinas se han mostrado capaces de reducir los eventos cardiovasculares17 e incluso estabilizar las placas de ateroma18. Sin embargo, la estabilización de estas placas se produce tras varios meses de tratamiento. La mayoría de los eventos cardiovasculares ocurren en las primeras semanas tras el episodio inicial19. Por tanto, hay una necesidad imperiosa de intervenciones que puedan reducir la vulnerabilidad de las placas de ateroma de manera aguda. Como se verá en secciones ulteriores, las HDL pueden tener un papel importante no sólo en la reducción del volumen de la placa, sino en la estabilización de ésta.
TRANSPORTE INVERSO DE COLESTEROL. BIOLOGÍA DE LAS HDL
Como ya se ha visto, la acumulación de lípidos en la pared arterial es un proceso dinámico y bidireccional en el que existen mecanismos naturales de extracción del colesterol acumulado en la pared del vaso.
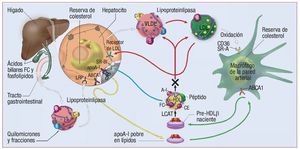
El transporte inverso de colesterol (TRC) se define como la extracción de colesterol de tejidos extrahepáticos y su movilización hacia el hígado para su metabolización y eventual excreción intestinal con los ácidos biliares. Como ya se ha explicado, las HDL tienen un papel central en la extracción de colesterol de las lesiones ateroscleróticas y en su transporte hasta el hígado para su ulterior metabolismo y final excreción intestinal para su eliminación junto con las heces. La figura 2 ilustra el papel crucial de las HDL en el TRC20.

Fig. 2. Esquema del transporte inverso de colesterol y protagonismo de las lipoproteínas de alta densidad (HDL) en él. La principal proteína de las HDL, la apolipoproteína (apo) A-I, la sintetizan el hígado y el intestino. Nada más entrar en el torrente circulatorio, se lipida (y se une a fosfolípidos que harán de reservorio para el transporte del colesterol). La apoA-I «guía» la HDL naciente (pobre en lípidos y proporcionalmente rica en proteína) hacia los tejidos extrahepáticos, fundamentalmente hacia los macrófagos, donde interacciona con el receptor ABCA-1 (del inglés ATP binding cassette transporter A-1) y extrae el colesterol de éstos. De vuelta en el torrente circulatorio, el LCAT (del inglés lecithin-cholesterol acyltransferase [transferasa de grupos acilo desde lecitina a colesterol]) esterifica el colesterol, y se forman las HDL maduras. Éstas pueden ir al hígado, donde interaccionan fundamentalmente con el receptor SR-BI de los hepatocitos y «vierten» el colesterol en el hígado para su ulterior excreción biliar, o intercambian su colesterol esterificado por triglicéridos con lipoproteínas de baja densidad (LDL) y muy baja densidad (VLDL), que podrán ir al hígado a eliminar el colesterol (vía receptor LDL) o de nuevo a los tejidos extrahepáticos. (Adaptado con permiso de Brewer et al20.) cHDL: colesterol de las lipoproteínas de alta densidad.
La principal proteína de las HDL es la apolipoproteína (apo) A-I, encargada del destino de las HDL. La apoA-I constituye más del 70% del contenido proteínico del total de partículas de HDL; de aquí que la concentración plasmática de ApoA-I, en condiciones normales (sin intervención farmacológica), se correlaciona estrechamente con la concentración plasmática de HDL. La apoA-II es la segunda apolipoproteína más abundante en las HDL, pero su misión todavía no ha sido bien definida. Las HDL contienen otras proteínas en menor concentración21,22. La importancia de la apoA-I en la biología de las HDL ha quedado patente en múltiples estudios básicos. La deleción del gen de apoA-I causa concentraciones extremadamente bajas de HDL en ratones23. De hecho, los ratones con predisposición a la aterosclerosis que carecen de apoA-I contraen una enfermedad aterosclerótica mucho más agresiva24. Por el contrario, la sobrexpresión hepática de apoA-I incrementa la concentración de HDL, inhibe la progresión e incluso consigue la regresión de la aterosclerosis en animales de experimentación25,26. Por ello se considera que la sobrexpresión endógena de apoA-I es una de las estrategias más prometedoras en las terapias relacionadas con el aumento de HDL. Sin embargo, estudios in vivo en humanos han mostrado que el principal factor que determina las concentraciones plasmáticas de HDL es la tasa de aclaramiento de apoA-I, y no su tasa de síntesis27.
A continuación se describen brevemente los pasos del TRC (fig. 2).
Síntesis de HDL
La síntesis y la secreción a la circulación de apoA-I (principal componente del HDL) se producen tanto en el hígado28 como en el intestino29; el hígado produce un 75% de la apoA-I humana30,31. Ambos tejidos se encargan de la lipidación de las moléculas recién secretadas de apoA-I pobre en lípidos, vía receptor ATP binding cassette A-1 (ABCA-1). Las HDL nacientes (que también reciben el nombre de apoA-I pobre en lípidos) generalmente contienen dos moléculas de apoA-I por partícula, mientras que los lípidos (fosfolípidos y colesterol libre) apenas suponen el 10% del total de su masa32.
Captación de colesterol por las HDL nacientes
Este proceso puede llevarse a cabo mediante un variado número de mecanismos33, y resulta finalmente en la formación de partículas discoidales de HDL:
- Difusión acuosa: la salida de colesterol libre por este mecanismo se lleva a cabo en todos los tipos celulares, pero es bastante ineficaz. Este mecanismo pasivo se cumple mediante un simple proceso de difusión, de modo que el movimiento de colesterol puede ser bidireccional, y el sentido del flujo está determinado únicamente por el gradiente químico de concentración de colesterol. Se trata, además, de un proceso lento (el paso de lípidos a través de la bicapa lipídica tarda horas).
- Salida de colesterol libre mediada por ABCA-1: contrariamente al mecanismo de difusión acuosa, este movimiento de colesterol libre a través de ABCA-1 es unidireccional, únicamente desde las células a las apoliproteínas pobres en lípidos.
- Scavenger receptor BI (SR-BI): de manera similar a la difusión, pero en claro contraste con ABCA-1, el flujo de colesterol libre mediado por la molécula de SR-BI tiene lugar sólo hacia aceptores que contengan fosfolípidos (es decir, HDL y apolipoproteínas lipidadas) y el flujo de colesterol libre es bidireccional (depende sólo del gradiente de concentración de colesterol libre a ambos lados de la membrana).
- ABCG-1/ABCG-4: se ha descubierto recientemente que la molécula ABCG-1 también supone una ruta alternativa para el transporte de colesterol libre desde los macrófagos hacia las HDL maduras34, nunca hacia HDL nacientes (apoA-I pobre en lípidos).
Maduración y remodelado de HDL
Las partículas de HDL nacientes sufren un proceso intravascular de remodelado y maduración35, mediante la acción de diversas enzimas:
- LCAT (lecitina-colesterol aciltransferasa): en el interior de la molécula discoidal de HDL naciente, LCAT cataliza la transferencia de grupos 2-acil desde la lecitina al colesterol libre captado desde los macrófagos, con lo que se generan ésteres de colesterol y lisolecitina36. Los ésteres de colesterol son más hidrófugos que el colesterol libre, por lo que se mueven al núcleo de la particula de lipoproteína, y así se forma la molécula madura de HDL, grande y esférica37. LCAT es esencial para el adecuado metabolismo de las HDL, dado que su ausencia impide la correcta formación de partículas maduras de HDL con sus núcleos lipídicos normales de ésteres de colesterol.
- CETP (proteína de transferencia de ésteres de colesterol): la CETP es una glucoproteína hidrófuga, sintetizada en el hígado y en el tejido adiposo, que circula unida a lipoproteínas en el plasma. La CETP promueve la transferencia de ésteres de colesterol de las partículas de HDL a las lipoproteínas que contengan la apoproteína B (no sólo LDL, sino también los quilomicrones y lipoproteínas de muy baja densidad [VLDL]) a cambio de triglicéridos, es decir, de manera inversa transfiere triglicéridos desde VLDL, quilomicrones y LDL a las HDL38,39, de manera que el resultado final es la migración de ésteres de colesterol de nuevo a LDL; el ciclo del TRC se completa con la captación de ésteres de colesterol de nuevo por el hígado gracias a los receptores de LDL hepáticos. El efecto total de la CETP en las HDL consiste en su depleción de ésteres de colesterol y su enriquecimiento en triglicéridos, de manera que se reduce el tamaño de la partícula de HDL. Por cierto, la inhibición de la CETP se ha propuesto como una terapia prometedora para el tratamiento de la aterosclerosis. Sin embargo, un reciente estudio en humanos para probar el efecto de un inhibidor de CETP (torcetrapib) resultó en un exceso de mortalidad en el grupo activo40.
- Otras proteínas implicadas son la proteína de transferencia de fosfolípidos y diferentes lipasas (lipoproteinlipasa, lipasa hepática y lipasa endotelial).
Catabolismo de las HDL
Como ya hemos comentado, el factor más determinante en las concentraciones plasmáticas de HDL y apoA-I es la tasa de aclaramiento de la apoA-I. Los riñones, el hígado y los tejidos productores de esteroides son los sitios principales de catabolismo de las HDL. Estudios en animales establecieron que un tercio de la apoA-I se cataboliza en los riñones, mientras que el resto se cataboliza en el hígado41. El aclaramiento de HDL puede tener lugar de dos maneras diferentes: a) captación de toda la partícula (holopartícula): endocitosis y degradación lisosómica de toda la partícula (incluida la apoA-I), que ocurren tanto en el hígado como en el riñón, y b) captación selectiva de colesterol: retirada del colesterol y de otros lípidos de la partícula, sin afectar al contenido proteínico. El mecanismo mejor caracterizado es la captación hepática por el receptor hepático SR-BI42. Se trata de un receptor que media la captación selectiva de otros lípidos, con constantes de captación más altas para ésteres de colesterol y colesterol libre y más bajas para fosfolípidos y triglicéridos.
Se ha postulado que el colesterol transportado en HDL se dirige más directamente hacia la excreción biliar que otros tipos de colesterol43. El colesterol libre movilizado en HDL puede excretarse directamente a la bilis o convertirse en ácidos biliares (la enzima limitante para esta reacción es la 7α-hidroxilasa) previamente a la excreción biliar32.
En resumen, el TRC es un mecanismo complejo mediante el que el colesterol de tejidos extrahepáticos es transportado hacia el hígado para su ulterior metabolismo y excreción por la bilis. Las HDL tienen un papel fundamental en el TRC, y existen algunos receptores clave en diferentes tejidos, de los que el ABCA-1 de los macrófagos y el SR-BI de los hepatocitos son receptores clave.
EFECTOS VASCULARES DE LAS HDL INDEPENDIENTES DEL TRC
Los efectos beneficiosos de las altas concentraciones de HDL se atribuyeron exclusivamente al efecto de éste en el TRC. Sin embargo, hoy se sabe que existen mecanismos no relacionados con el TRC que contribuyen al efecto vasculoprotector de las HDL. Las HDL, y más concretamente su principal proteína (apoA-I), tienen efectos antioxidantes directos. En consecuencia, HDL/apoA-I tienen la capacidad de inhibir la oxidación de las LDL (mecanismo capital en la aterogénesis, como ya se ha expuesto). Las HDL, además, tienen otras propiedades antiinflamatorias, como la inhibición de la expresión de moléculas de adhesión en células endoteliales. Esta inhibición redunda en una disminución del reclutamiento de monocitos circulantes por la pared vascular. Para más detalle sobre las propiedades antiinflamatorias de HDL/apoA-I, se puede consultar textos extensos al respecto44,45.
Debido a que las acciones beneficiosas de las HDL implican todos los procesos relacionados con el desarrollo, la progresión y la aparición de síntomas de la aterosclerosis, el aumento de HDL puede ser un objetivo muy pertinente para el tratamiento de esta enfermedad.
NUEVAS ESTRATEGIAS PARA INCREMENTAR LAS HDL
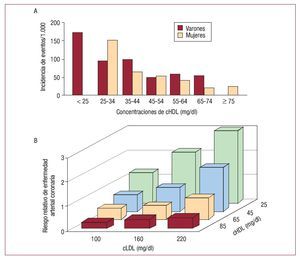
Se ha comprobado de manera fehaciente que la reducción de LDL es una estrategia efectiva y segura para reducir el riesgo de enfermedad cardiovascular3. A pesar de los impresionantes beneficios que se han demostrado con las estatinas en reducir la morbimortalidad, todavía existe una proporción nada desdeñable de pacientes con concentraciones de LDL consideradas óptimas que siguen sufriendo eventos cardiovasculares. Por esta razón es evidente que hay necesidad imperiosa de reducir el riesgo residual de eventos cardiovasculares. No sólo se sabe que las bajas concentraciones de HDL son un importante factor independiente de riesgo de enfermedad cardiovascular46, sino que se ha demostrado que el efecto vasculoprotector de las altas concentraciones de HDL se mantiene para todo el espectro de concentraciones de LDL, incluso para los pacientes con concentraciones bajas de LDL47-49 (fig. 3). Por todo ello, la idea de incrementar las concentraciones de HDL representa una estrategia moderna, novedosa y prometedora en la aterotrombosis.

Fig. 3. Concentraciones de colesterol de las lipoproteínas de alta densidad (cHDL) y su correlación con eventos cardiovasculares en el estudio de Framingham. Como puede verse, la correlación inversa entre cHDL y eventos cardiovasculares se mantiene para cualquier concentración de colesterol de las lipoproteínas de baja densidad (cLDL), incluso las más bajas. Esto indica un beneficio adicional (quizá independiente) al de la disminución de éstas. (Adaptado con permiso de Genest et al49.)
Basándose en la relación inversamente proporcional entre concentraciones de HDL y enfermedad cardiovascular, Miller et al50 postularon por primera vez que un incremento de las HDL representaría una nueva frontera para el tratamiento de la aterosclerosis. En esta línea, el primer estudio de intervención fue realizado por nuestro grupo a finales de los años ochenta. Nosotros demostramos por primera vez que la administración exógena de HDL era capaz de hacer que remitieran la lesiones ateromatosas generadas en un modelo animal51,52. Desde esta primera demostración experimental del efecto beneficioso de las HDL en la aterosclerosis51-53, éstas se convirtieron en materia de alto interés científico y clínico.
Existen diferentes abordajes de intervención para el aumento de las HDL. Además de una modificación de los hábitos de vida (realización de ejercicio, consumo moderado de alcohol, etc.), el aumento de las HDL circulantes puede conseguirse principalmente mediante diferentes dianas terapéuticas: aumento de síntesis de la apoA-I y/o disminución de su catabolismo, inhibición de la CETP e infusión de HDL/apoA-I exógenas, como más representativas. Un aumento en la síntesis hepática de la apoA-I puede conseguirse con agonistas de los receptores activados por proliferadores de peroxisomas (PPARα), como los fibratos. Además de disminuir de forma importante los triglicéridos y de forma modesta las LDL, los fibratos aumentan significativamente la concentración de HDL. Diferentes ensayos clínicos han estudiado el efecto de los fibratos en la prevención de eventos cardiovasculares, con resultados esperanzadores aunque no definitivos54,55. Estudios en marcha deberán poner en perspectiva real el efecto de esta terapia en pacientes ya tratados con estatinas.
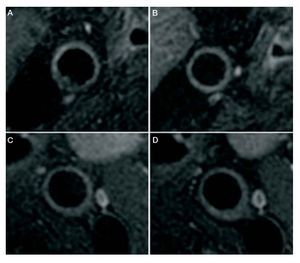
El ácido nicotínico (niacina) y sus derivados aumentan de forma muy importante la concentración de HDL y disminuyen el catabolismo de la apoA-I. Estudios piloto han indicado su posible eficacia en la regresión de placas de ateroma56. Muy recientemente, el grupo de Choudhury ha publicado el primer estudio que analiza el efecto antiaterosclerótico de la niacina en pacientes en tratamiento con estatinas y concentraciones de LDL muy bajas (en torno a 80 mg/dl)57. Por primera vez se ha demostrado que el incremento de HDL mediante tratamiento con niacina es capaz de reducir el volumen de placa de ateroma carotídea evaluado mediante resonancia magnética (fig. 4).

Fig. 4. Regresión de placa de ateroma en carótida de pacientes tratados con niacina (ácido nicotínico). Imágenes de resonancia magnética de cortes transversales de carótidas antes (A y C) y después de 12 meses de tratamiento con niacina (B y D). Puede verse una regresión del volumen de placa en el grupo con niacina (A y B) y ausencia de progresión en el grupo con placebo (C y D). Ambos grupos estaban tratados con estatinas y tenían concentraciones de lipoproteínas de baja densidad acordes con las guías de actuación clínica. (Reproducido con permiso de Lee et al57.)
Sin embargo, hasta la fecha, la alta tasa de efectos secundarios menores (principalmente rubefacción) que conlleva el tratamiento con niacina ha hecho difícil la validación en grandes estudios clínicos. Recientemente se ha comercializado un nuevo fármaco que combina niacina con un inhibidor del rubor facial y es una de las terapias más novedosas para aumentar las HDL en la enfermedad aterotrombótica58.
Otro grupo de fármacos que aumentan de forma significativa las HDL son los inhibidores de la CETP. Estudios en animales de experimentación demostraron que la inhibición de la CETP resultaba en un efecto beneficioso antiaterosclerótico59. Después de estos estudios preclínicos, estudios piloto en humanos corroboraron un aumento de HDL de un 35-50% con la administración de inhibidores de la CETP60-62. Tras estos resultados tan esperanzadores, se estudió el efecto del inhibidor de la CETP torcetrapib en un gran ensayo multicéntrico (ILLUMINATE). Sorprendentemente, el estudio tuvo que suspenderse de manera prematura por un aumento en la mortalidad de los pacientes en el brazo de torcetrapib, pese a un aumento muy significativo de las HDL40. La causa de este fracaso no se conoce con certidumbre, pero la hipótesis más barajada es que pudo deberse a un fallo de la molécula (toxicidad vascular directa, hipertensión e interacción con el sistema renina-angiotensina-aldosterona del torcetrapib)63, y no la inhibición de la CETP en sí misma. Estudios en marcha analizan el efecto de otros inhibidores de la CETP64,65.
La infusión exógena de HDL y derivados ha sido estudiada por diferentes grupos, tras nuestra primera evidencia en un modelo de conejo hace ya 20 años52. El tipo de HDL del que se tiene más experiencia preclínica es la infusión de HDL recombinante formada por la apoA-IMilano y fosfolípidos (HDLMilano). La infusión de HDLMilano se ha estudiado en un ensayo piloto en humanos, con resultados prometedores66. Otra forma de HDL estudiada en humanos —con resultados iniciales prometedores— es la HDL reconstituida que contiene apoA-I nativa humana y fosfolípidos67. Ambas formas sintéticas de HDL se han estudiado en infusiones intravenosas de corta duración (una infusión semanal durante aproximadamente 1 mes), y se han obtenido resultados significativos en cuanto a regresión de placa a nivel coronario en pacientes con aterotrombosis clínica. La principal ventaja potencial de estas formas de HDL es la rapidez de su efecto en reducción de placa.
HDL RECOMBINANTE (apoA-IMILANO):
HDLMILANO
La HDLMilano es el resultado de una mutación espontánea de la apoA-I (conocida como apoA-IMilano), identificada por el grupo de Sirtori a finales de los años setenta en un pequeño pueblo cercano a Milán llamado Limone Sul Garda68,69. Una familia de este pueblo se mostró resistente a la aterosclerosis pese a tener un perfil lipídico proaterogénico consistente en bajas concentraciones de HDL y apoA-I junto con elevadas concentraciones de triglicéridos. Estudios de electroforesis demostraron que estos individuos tenían una mutación de la apoA-I resultado de la sustitución de un único aminoácido de la proteína apoA-I. Esta proteína está compuesta de 273 aminoácidos, y la mutación Milano consiste en la sustitución Arg por Cis en posición 173. Los sujetos con esta mutación de HDL tienen una tasa muy baja de aterosclerosis tanto clínica como subclínica70. La baja tasa de eventos cardiovasculares de estos sujetos motivó la teoría de que esta HDL mutante, que contiene apoA-IMilano, sea mucho más funcional que la apoA-Inativa70. La proteína mutante apoA-IMilano ha sido clonada y un laboratorio la ha unido al fosfolípido palmitoil-2-oleoil fosfatidilcolina (POPC) formando HDL sintética (HDL recombinante [apoAIMilano]). Este tipo de HDL sintética ha sido estudiado por diferentes grupos en distintos modelos experimentales, con unos resultados muy homogéneos en cuanto a regresión de la placa de ateroma71-77. Además de múltiples estudios preclínicos, se ha realizado un estudio piloto en pacientes con síndrome coronario agudo, que corroborado la regresión aguda de ateroma coronario66.
Por lo tanto, hay evidencia indiscutible de que la infusión exógena de HDLMilano es capaz de disminuir el volumen de placa tanto en modelos animales como en humanos. Como se ha descrito previamente, las manifestaciones clínicas de la aterotrombosis están dictadas en gran parte por la composición de la placa (vulnerabilidad), y no por la presencia de ateroma más o menos estenótico. Recientemene, nosotros hemos publicado un estudio en animales de experimentación en el que evaluamos el efecto de la infusión aguda de HDLMilano en el volumen y la composición de las placas de ateroma78. Confirmando resultados contemporáneos de otros grupos independientes79, la administración aguda de HDLMilano indujo una regresión muy significativa del volumen de placa de ateroma (fig. 5). Este efecto fue evaluado tanto con resonancias magnéticas seriadas como con estudios de tomografía computarizada (TC) multicorte, y se probó que esta ténica de imagen también es capaz de cuantificar los cambios evolutivos de la carga aterosclerótica80, por lo que sería una herramienta muy prometedora para la evaluación no invasiva de terapias antiaterogénicas81. Es interesante que esta regresión de la placa se viera acompañada de un cambio cualitativo, lo que indica una estabilización aguda de la placa de ateroma78. Hasta la fecha, éste es el primer estudio que correlaciona la regresión de la placa de ateroma in vivo con cambios en la vulnerabilidad de la placa tras la administración de HDLMilano. El mecanismo de acción relacionado con estos efectos beneficiosos tan agudos parece ser tanto a un incremento muy importante del TRC como un efecto antiinflamatorio generalizado que promueve un «rescate» del estado inflamatorio general que la enfermedad aterotrombótica conlleva82. Merece destacarse el hecho de que todos estos efectos beneficiosos en el volumen y la estabilidad de la placa se vieran tras sólo dos infusiones de HDLMilano (en 4 días). Actualmente existen diferentes intervenciones que se han demostrado eficaces en la estabilización de la placa de ateroma83; sin embargo, el tiempo necesario para inducir tal efecto fue relativamente largo (varios meses). Es importante enfatizar que la mayoría de los eventos recurrentes ocurren en la fase aguda tras el primer evento isquémico19. Nuestros resultados indican que una dosis de carga intravenosa de HDLMilano puede inducir una acelerada regresión de placa, con un efecto estabilizador de las placas activas y de alto riesgo.

Fig. 5. Regresión de placa de ateroma tras tratamiento agudo con HDLMilano. Imágenes de resonancia magnética de cortes transversales de aorta abdominal de conejos ateroscleróticos antes y después de tratamiento con HDLMilano o placebo. HDLMilano resultó en una regresión muy significativa del volumen de placa, mientras que el placebo no tuvo efecto. Es importante enfatizar que esta regresión de placa se vio tras únicamente dos infusiones de HDLMilano (75 mg de apoA-IMilano/kg). Además de una regresión brusca del volumen de placa, estudios de expresión génica y proteínica mostraron signos importantes de estabilización de la placa. (Reproducido con permiso de Ibáñez et al78.)
CONCLUSIONES
Las altas concentraciones séricas de HDL se han correlacionado con una disminución de la incidencia de eventos aterotrombóticos; por lo tanto, la posibilidad de incrementar las HDL con fármacos es un abordaje perseguido para luchar contra esta enfermedad epidémica. Existen diferentes dianas terapéuticas para conseguir un aumento significativo de las HDL circulantes. Se ha estudiado diferentes grupos de fármacos, con resultados prometedores en tratamientos de larga duración. El beneficio de incrementar las HDL parece ser adicional al relacionado con el tratamiento crónico con estatinas. La infusión intravenosa de diferentes formas de HDL reconstituida resulta en una regresión muy aguda de la placa, que se asocia con una estabilización de la placa en el caso de HDLMilano. A la espera de la confirmación de estudios de eventos cardiovasculares, la terapia dirigida a aumentar las HDL es una frontera cercana que puede ser un paso adelante en el tratamiento de la aterotrombosis.
Full English text available from: www.revespcardiol.org
Correspondencia: Dr. J.J. Badimón.
Atherothrombosis Research Unit. The Zena and Michael A. Wiener Cardiovascular Institute. Mount Sinai School of Medicine.
1 Gustave Levy Place. Box 1030. New York, NY 10029. Estados Unidos.
Correo electrónico: juan.badimon@mssm.edu