Keywords
INTRODUCTION
Atherosclerosis can be defined in very simplified and reductionist terms as the result of the imbalance between cholesterol entering and leaving the arterial wall in such a way that entry predominates. As Figure 1 shows, the main factor involved in cholesterol entering the arterial wall is low-density lipoprotein (LDL), whereas the main factor governing cholesterol leaving the arterial vessel is high-density lipoprotein (HDL). Both high systemic (bloodstream) concentrations of LDL cholesterol (LDL-C) and low concentrations of HDL cholesterol (HDL-C) have been constantly associated with the development of atherosclerosis. Following this discovery, multiple therapies have been employed in the attempt to prevent the development of the disease by reducing LDL or increasing HDL. Although reducing LDL, mainly by using statins, is still the established standard therapy for the primary and secondary prevention of atherothrombotic events, it still has not been conclusively shown that increasing HDL provides benefit. In this article, we review and place in historical perspective the biology of HDL, the interventions available that increase it, and their potential benefits.

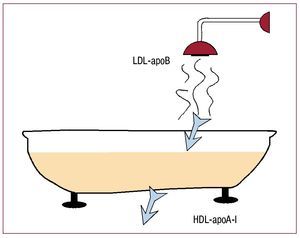
Figure 1. Pictorial representation of the main agents involved in the influx and efflux of cholesterol. The bathtub represents the vessel wall. The main agents controlling cholesterol entering the vessel are low-density lipoproteins (LDL), guided by their main protein, apolipoprotein (apo) B. High-density lipoproteins (HDL), on the other hand, control cholesterol efflux from the arterial wall by the interaction between their main protein, ApoA-I, and the receptors in the macrophage.
PATHOPHYSIOLOGY OF ATHEROSCLEROSIS
The association between a fat-rich diet (cholesterol) and atherosclerotic disease was accepted many years ago. However, it was not until the beginning of the 20th century that Ignatowski demonstrated it scientifically for the first time.1 This author was able to generate atherosclerotic vascular lesions in rabbits by providing them with a diet rich in milk and egg yolk. Another Russian scientist, Nikolai Anitschkow, identified cholesterol as the dietary component that causes the development of atherosclerotic lesions.2
Atherothrombotic disease is the leading cause of mortality in western countries and is becoming the first in developing countries. Thus, research into atherosclerosis is fundamental to check the spread of this global epidemic.
Atherosclerosis is a systemic disease with local clinical manifestations3 and it is mainly characterized by lipid deposition on the walls of medium- and large-sized arteries. High concentrations of circulating LDL-C lead to it accumulate in the arterial intima. These small particles of LDL are deposited in the areas of the intima that are rich in proteoglycans and converge into aggregates of LDL-C. The binding of LDL-C particles to proteoglycans "sequesters" LDL-C, which prolongs contact between LDL-C and the arterial intima.4 This binding process makes LDL-C particles more susceptible to oxidation. The oxidation of LDL-C is considered a key event in the pathogenesis of atherosclerosis.5 Prolonged contact of oxidized LDL-C to the arterial intima, as well as exposure to different risk factors (hypertension, smoking, diabetes mellitus, etc), can lead to endothelial dysfunction,6 which is the earliest pathological process of atherosclerosis.7 Once the endothelium is damaged, LDL-C infiltrates the deepest arterial layers. In an attempt to check this LDL-C "invasion," circulating monocytes and macrophages penetrate the arterial wall, phagocytize the oxidized LDL-C, and become cholesterol-loaded cells. When the macrophages are completely loaded with cholesterol, they become foam cells. Since cholesterol is not easily metabolized outside the liver, its continuous accumulation within the cells, which deteriorate, leads to apoptosis, with the subsequent release of prothrombotic substances, such as tissue factor.8 After the foam-macrophage cells die, the cholesterol is again released into the arterial wall forming the lipid core of the plaque, thus perpetuating the process.9 Furthermore, cholesterol can crystallize, and this has recently been identified as a factor in the destabilization of plaque.10
Apart from lipid accumulation, the recruitment of additional inflammatory cells to monocytes and macrophages is an important factor in atherogenesis. Thus, the cellularity of the atherosclerotic lesions is modified as the disease progresses. The initial lesions are fatty streaks that mainly carry cholesterol-loaded macrophages and some leukocyte cells. As the disease progresses, the lesion becomes more complex, with different cell types and a great deal of extracellular material. As the lipid core of the atheromatous plaque grows by the accumulation of LDL-C particles and macrophages, smooth muscle cells migrate from the media to the intima. These cells produce and secrete collagen and fibrous elements of the extracellular matrix, leading to the formation of the fibrous covering of the fibroatheromatous plaques.
At first, these plaques do not reduce the vascular lumen since there is compensatory dilatation of the vascular wall (positive remodelling).11 The plaque grows eccentrically, leading to thinning of the media and the adventitia, until compensatory dilatation cannot continue, from which time it begins to grow toward the center of the vascular lumen, thus compromising blood flow.
The majority of acute cardiovascular events are not caused by progressive narrowing of the vascular lumen, but by complications arising from atherosclerotic plaque (rupture, ulceration, hemorrhage, erosion) that lead to acute vascular occlusion due to vessel thrombosis. Plaques with a high lipid content have also been called "unstable," "vulnerable," or "high-risk," due to their propensity to rupture and increase the possibility of an acute event. From the clinical point of view, atherosclerosis is basically characterized by the formation of an acute (occlusive) thrombus anchored to the broken or eroded plaque, and thus the term atherothrombosis more accurately describes the nature of the disease.
ATHEROTHROMBOSIS: IMPORTANCE OF PLAQUE COMPOSITION
"Man lives with atherosclerosis, but dies of thrombosis."
Although lipid accumulation on the arterial walls is the basis of atherosclerosis, the mere presence of atheromatous plaques does not generate any symptoms as such. The clinical manifestations of the disease are secondary to rupture of atheromatous plaques with superimposed thrombosis. Depending on to what extent the thrombus impedes arterial flow, the manifestations will be more severe or less severe. Thrombosis is the fundamental mechanism of transition from a chronic-latent state of disease to a symptomatic acute state.
The plaques traditionally considered as high-risk are usually those with abundant lipid components, separated from the vessel lumen by a thin layer of extracellular matrix covered by the endothelium.12 The rupture of atheromatous plaque seems to be driven by some passive mechanisms (shearing stress) and active mechanisms (macrophage infiltration and activation, proteolytic enzyme release, inflammatory cell infiltration, etc). The presence of matrix metalloproteinases (MMPs) is of special interest. The MMPs are enzymes that degrade the extracellular matrix of the plaque, making it more unstable and therefore more prone to rupture. It has been observed that, in atherosclerotic lesions, MMPs are expressed in the regions with a greater propensity to rupture, where they are found together with macrophages, and this confirms their involvement in plaque instability.13
The local and systemic mechanisms that modulate thrombosis are highly relevant in this disease. The factors external to the plaque (systemic factors) also modulate its vulnerability, as well as thrombogenicity once it has ruptured. In fact, some systemic factors, such as high LDL-C concentrations, smoking, hyperglycemia, diabetes and others, are associated with an increase in the coagulability of the blood.14 Some recent studies have demonstrated that the endothelial cells that cover the atherosclerotic plaques can become procoagulant due to apoptosis induction.15
In recent decades, the relevance of inflammation in atherothrombosis has been highlighted. In fact, the presence of local and systemic inflammation markers is correlated with increased patient vulnerability (ie, the risk of suffering an atherothrombotic event).16
All the foregoing leads to the conclusion that the vulnerability of atheromatous plaques is modulated by local and systemic factors. The vulnerability of the plaques can be locally and systemically reduced (ie, stabilized). Atheromatous plaque stabilization can be achieved by the use of drugs. Statins have demonstrated capacity to reduce cardiovascular events17 and to even stabilize atheromatous plaques.18 However, the stabilization of such plaques only occurs after several months of treatment . Most cardiovascular events occur in the first weeks after the initial episode.19 Therefore, there is a pressing need for interventions that can lead to acute reductions in the vulnerability of atheromatous plaques. As shown in the following sections, HDL may play an important role not only in the reduction of plaque volume, but in its stabilization.
REVERSE CHOLESTEROL TRANSPORT. THE BIOLOGY OF HIGH-DENSITY LIPOPROTEIN
As mentioned, lipid accumulation on the arterial wall is a dynamic and bidirectional process involving natural mechanisms that extract the cholesterol that has accumulated on the vessel wall.
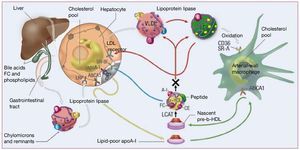
Reverse cholesterol transport (RCT) is defined as the efflux of cholesterol from extrahepatic tissue and its transport to the liver for metabolization and ultimately excretion via the intestines together with bile acids. As described, HDL plays a key role in the efflux of cholesterol from atherosclerotic lesions and its transport to the liver for metabolization and final excretion via the intestines together with the feces. Figure 2 illustrates the key role of HDL in RCT.20

Figure 2. Reverse cholesterol transport and the role of high-density lipoproteins (HDL). The main protein of HDL, apolipoprotein A-I (ApoA-I), is synthesized by the liver and intestine. As it enters the bloodstream, it is lipidized (and bonds with phospholipids that will act as a reservoir for cholesterol transport ). ApoA-I "guides" nascent HDL (poor in lipids and rich in protein) toward the extrahepatic tissues, mainly toward macrophages, where it interacts with the ATP binding cassette transporter A-1 (ABCA-1) receptor, and extracts cholesterol from them. Back in the bloodstream, lecithin-cholesterol acyltransferase (LCAT) esterifies the cholesterol, and mature HDL is formed. This can return to the liver, where they mainly interact with the SR-BI receptors in the hepatocytes and "pour" the cholesterol into the liver for subsequent biliary excretion, or can exchange their esterified cholesterol for triglycerides with low-density lipoproteins (LDL) and very-low-density (VLDL), that can travel to the liver to eliminate the cholesterol (via the LDL receptor) or once again to the extrahepatic tissues. (Adapted with permission from Brewer et al20) HDL-C indicates high-density lipoprotein cholesterol.
The main HDL protein is apolipoprotein A-I (ApoA-I), whose role is the transportation of HDL. ApoA-I constitutes more than 70% of the total protein content in HDL particles; for this reason, in normal conditions (ie, without pharmacological intervention), the ApoA-I plasma concentrations are closely correlated with HDL plasma concentrations. Apolipoprotein A-II is the second most abundant apolipoprotein in HDL, but its role has yet to be elucidated. High-density lipoprotein contains some other proteins in lower concentrations.21,22 The importance of ApoA-I in the biology of HDL has been clarified in several basic research studies. The deletion of the ApoA-I gene causes extremely low concentrations of HDL in mice.23 In fact, mice with a predisposition to atherosclerosis that lack ApoA-I develop much more aggressive atherosclerotic disease.24 On the other hand, the hepatic overexpression of ApoA-I increases HDL concentrations, inhibits atherosclerosis progression, and even causes the regression of atherosclerosis in laboratory animals.25,26 Thus, endogenous ApoA-I overexpression is one of the most promising strategies in therapies aimed at increasing HDL concentrations. However, in vivo studies in humans have shown that the main factor affecting plasma HDL concentrations is the ApoA-I clearance rate, and not the ApoA-I synthesis rate.27
The steps involved in RCT are briefly described below (Figure 2).
Synthesis of HDL
The synthesis and secretion of ApoA-I (the main component of HDL) in the bloodstream take place in the liver28 and in the intestine29; the liver produces 75% of human ApoA-I.30,31 Both these types of tissues control the lipidation of freshly secreted lipid-poor ApoA-I, via the ATP binding cassette transporter A-1 (ABCA-1) receptor. The nascent HDL (which is also called lipid-poor ApoA-I) usually contains 2 molecules of ApoA-I per particle, whereas the lipids (phospholipids and free cholesterol) barely form 10% of their total mass.32
Cholesterol Uptake by Nascent HDL
This process takes place via a number of mechanisms,33 which finally results in the formation of discoidal particles of HDL:
- Aqueous diffusion: the efflux of free cholesterol by this mechanism takes place in all cell types, but is quite inefficient. This passive mechanism involves a simple diffusion process whereby cholesterol movement can be bidirectional, and in which the direction of the flow only depends on the cholesterol concentration gradient. In addition, it is a slow process (the passage of lipids through the lipid bilayer takes several hours).
- ABCA-1-mediated free cholesterol efflux: in contrast to the aqueous diffusion mechanism, the ABCA-1-mediated movement of free cholesterol is unidirectional, that is, from the cells to lipid-poor apoliprotein.
- Scavenger receptor class-B type I (SR-BI): as in the diffusion process, but in clear contrast to ABCA-1, the flux of free cholesterol mediated by SR-BI only takes place toward phospholipid-containing acceptors (ie, HDL and lipidated apolipoprotein) and the flux of free cholesterol is bidirectional (dependent only on the free cholesterol concentration gradient on both sides of the membrane).
- ABCG-1/ABCG-4: it was recently discovered that ABCG-1 also provides alternative pathway for the transport of free cholesterol from macrophages to mature HDL,34 but never toward nascent HDL (lipid-poor ApoA-I).
Maturation and Remodeling of HDL
The nascent HDL particles undergo an intravascular process of remodeling and maturation35 by the action of several enzymes:
- LCAT (lecithin-cholesterol acyltransferase): within the discoidal nascent HDL molecule, LCAT catalyzes the transfer of 2-acyl groups from lecithin to free cholesterol from macrophages, and in this way cholesterol esters and lysolecithin are generated.36 The cholesterol esters are more hydrophobic than free cholesterol, and therefore they move to the core of the lipoprotein particle, thus forming a mature HDL molecule, which is large and spherical.37 Lecithincholesterol acyltransferase is essential for the correct metabolism of HDL, since its absence prevents the correct formation of mature HDL particles with normal lipid cores of cholesterol esters.
- CETP (cholesterol ester transfer protein): the CETP is a hydrophobic glycoprotein synthesized in the liver and adipose tissue, and that circulates bound to lipoproteins in plasma. Cholesterol ester transfer protein promotes the transfer of cholesterol esters from HDL particles to lipoproteins that contain apoprotein B (not only LDL, but also chylomicrons and very-low-density lipoproteins [VLDL]) in exchange for triglycerides. That is, we can say that CETP transfers triglycerides from VLDL, chylomicrons and LDL to HDL,38,39 and therefore the end result is the migration of cholesterol esters back to LDL; the RCT cycle is completed with the re-uptake of cholesterol esters by the liver LDL receptors. The overall effect of CETP on HDL consists in depleting it of cholesterol esters and providing triglycerides, such that the size of the HDL particle is reduced. The inhibition of CETP has been suggested as a promising therapy for the treatment of atherosclerosis. However, a recent study to test the effect of a CETP inhibitor (torcetrapib) in humans resulted in increased risk of death in the active group.40
- Other proteins implicated in these processes include phospholipid transfer protein and several lipases (lipoprotein lipase, hepatic lipase and endothelial lipase).
HDL Catabolism
As mentioned, the key factor determining HDL and ApoA-I plasma concentrations is the ApoA-I clearance rate. The kidneys, liver, and steroid-producing tissues are the main sites for HDL catabolism. Animal studies have established that one-third of ApoA-I is catabolized by the kidneys, whereas the rest is catabolized in the liver.41 The clearance of HDL can take place in 2 different ways: a) uptake of the whole particle (holoparticle): endocytosis and lysosomal degradation of the whole particle (including ApoA-I), which occurs in the liver and in the kidney; and b) selective cholesterol uptake: removal of cholesterol and other lipids from the particle without affecting the protein content. The mechanism that has been best characterized is liver uptake by the hepatic SR-BI receptor.42 This receptor mediates the selective uptake of other lipids, with higher uptake for cholesterol esters and free cholesterol and lower uptake for phospholipids and triglycerides.
It has been suggested that the cholesterol transported in HDL is directed toward biliary excretion more than other types of cholesterol.43 The free cholesterol transported in HDL can be excreted directly to bile or can be converted into bile acids (the rate-limiting enzyme for this reaction is 7a hydroxylase) before bile excretion.32
In short, RCT is a complex mechanism enabling cholesterol from extrahepatic tissue to be transported toward the liver for its final metabolism and its excretion by bile. High-density lipoproteins play a key role in RTC, and there are key receptors in different tissues, with ABCA-1 in macrophages and SR-BI in hepatocytes being the most important.
VASCULAR EFFECTS OF REVERSE CHOLESTEROL TRANSPORT- INDEPENDENT HDL
The beneficial effects of high HDL concentrations used to be exclusively attributed to the effect of HDL on RCT. However, nowadays, it is known that there are mechanisms unrelated to RCT that also contribute to the vascular protective effect of HDL. High-density lipoprotein, and more specifically, its main protein (ApoA-I), have direct antioxidant effects. Thus, HDL/ ApoA-I have the capacity to inhibit LDL oxidation (the main mechanism in atherogenesis, as described). In addition, HDL has other antiinflammatory properties, such as inhibiting the expression of cell adhesion molecules in endothelial cells. This inhibition results in a reduction of the recruitment of circulating monocytes by the vascular wall. For more detail on the antiinflammatory properties of HDL/ ApoA-I, we refer the reader to some comprehensive studies.44,45
Since the beneficial activity of HDL involves all the processes associated with the development, progression, and onset of atherosclerosis symptoms, increasing the HDL concentrations could be a core target in the treatment of this disease.
NEW STRATEGIES TO INCREASE HDL
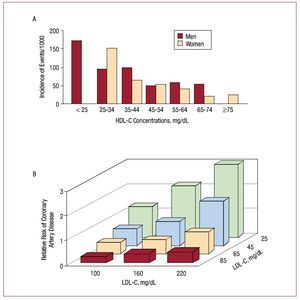
It has been demonstrated beyond doubt that reducing LDL is an effective and safe strategy to reduce the risk of cardiovascular disease.3 Despite the impressive benefits demonstrated by statins in reducing morbidity and mortality, there is still a considerable proportion of patients with optimal LDL concentrations who continue to suffer cardiovascular events. For this reason, there is a pressing need to reduce the residual risk of cardiovascular events. Not only is it known that low HDL concentrations are a relevant independent risk factor for cardiovascular disease, 46 but it has been demonstrated that the vascular protective effect of high HDL concentrations applies to the whole spectrum of LDL concentrations, even in patients with low LDL concentrations (Figure 3).47-49 Bearing this in mind, increasing the HDL concentrations is a modern, innovative and promising strategy in atherothrombosis.

Figure 3. High-density lipoprotein cholesterol (HDL-C) concentrations and their correlation with cardiovascular events in the Framingham study. The inverse correlation between HDL-C and cardiovascular events is maintained for any concentration of low-density lipoprotein cholesterol (LDL-C), including the lowest concentrations. This represents an extra benefit (perhaps independent) additional to reducing them. (Adapted with permission from Genest et al49).
Based on the inversely proportional association between HDL concentrations and cardiovascular disease, Miller et al50 were the first to suggest that increasing the HDL concentrations would represent a new frontier in the treatment of atherosclerosis. In this line, the first intervention study was conducted by our group at the end of the 1980s. We demonstrated for the first time that the exogenous administration of HDL achieves remission of atheromatous lesions generated in an animal model.51,52 Since first experimental demonstration of the beneficial effect of HDL on atherosclerosis,51-53 HDL has become a focus of great scientific and clinical interest.
There are different intervention approaches to increasing the HDL concentrations. In addition to life-style changes (more exercise, moderate alcohol consumption, etc), an increase of circulating HDL can be achieved via several therapeutic targets, among which the following are the most representative: increasing the synthesis of ApoA-I or reducing its catabolism, inhibiting CETP, and infusing exogenous HDL/ApoA-I. The hepatic synthesis of ApoA-I can be increased with peroxisome proliferator-activated receptors (PPARa), such as the fibrates. Apart from obtaining an important reduction of triglycerides and a moderate reduction of LDL, fibrates significantly increase HDL concentrations. Several clinical trials have studied the effect of fibrates on the prevention of cardiovascular events, and have obtained encouraging, but not definitive, results.54,55 Some ongoing studies may lead to more concrete results on the effect of this therapy on patients already receiving statin treatment.
Nicotinic acid (niacin) and its derivatives strongly increase HDL concentrations and reduce the catabolism of ApoA-I. Pilot studies have shown their potential efficacy in the regression of atheromatous plaques.56
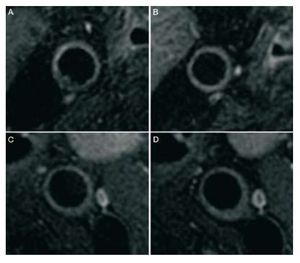
Very recently, the Choudhury group published the first study to analyze the anti-atherosclerotic effect of niacin on patients with very low LDL concentrations (around 80 mg/dL) who were being treated with statins.57 For the first time, it has been demonstrated that increasing the HDL concentrations via treatment with niacin is capable of reducing the volume of atheroma plaque in the carotid artery, as assessed by magnetic resonance imaging (Figure 4).

Figure 4. Atheromatous plaque regression in a carotid artery in patients treated with niacin (nicotinic acid). Magnetic resonance imaging of cross-sectional view of the carotid arteries before (A and C) and after 12 months of treatment with niacin (B and D). Regression of plaque volume in the niacin group (A and B) and the absence of progression in the placebo group (C and D) can be observed Both groups presented low-density lipoprotein concentrations and were treated with statins according to the clinical guidelines. (Reproduced with permission from Reads et al57).
However, up to the present, the high rate of minor adverse effects (mainly rubor) involved in treatment with niacin has made it difficult to validate it in large clinical studies. Recently, a new drug combining niacin with a facial rubor inhibitor has been marketed and is one of most innovative therapies to increase HDL in patients with atherothrombotic disease.58
The CETP inhibitors are another group of drugs that significantly increase HDL concentrations. Experimental animal studies have demonstrated that the CETP inhibition has a beneficial antisclerotic effect.59 After these preclinical studies, pilot studies in humans have corroborated an increase in HDL concentrations ranging between 35% and 50% after the administration of CETP inhibitors. 60-62 Following these encouraging results, the effect of the CETP inhibitor torcetrapib was studied in a large multicenter trial (ILLUMINATE). Surprisingly, the study had to be prematurely interrupted due to an increase in death among patients in the torcetrapib arm, despite a very significant increase in HDL concentrations.40 The cause of this failure is not known with any certainty, but the most popular hypothesis is that it might be due to the off-target adverse effects of the torcetrapib molecule (direct vascular toxicity, hypertension and interaction between the renin-angiotensin-aldosterone system and torcetrapib),63 and not to CETP inhibition itself. Other ongoing studies are analyzing the effect of other CETP inhibitors.64,65
Following the first results obtained in a rabbit model some 20 years ago,52 the exogenous infusion of HDL and derivatives has been studied by different groups. The type of HDL for which there is more preclinical experience is the infusion of recombinant HDL formed by ApoA-IMilano and phospholipids (HDLMilano). The infusion of HDLMilano was studied in a pilot trial in
humans, with promising results.66 Another form of HDL studied in humans—also with promising initial results—is reconstituted HDL containing native human ApoA-I and phospholipids.67 Both forms of synthetic HDL have been analyzed using short-term intravenous infusions (a weekly infusion for approximately 1 month), and significant results were obtained regarding the regression of plaque at the coronary level in patients with clinical atherothrombosis. The main potential advantage of these forms of HDL is their speed of effect in reducing plaque.
Recombinant HDL (ApoA-IMilano): HDLMilano
HDLMilano is the result of a spontaneous mutation of ApoA-I (known as ApoA-IMilano) identified by the Sirtori group in the late 1970s in Limone Garda Sul, a small town close to Milan.68,69 A family in the village seemed to be resistant to atherosclerosis despite having a proatherogenic lipid profile with low HDL and ApoA-I concentrations together with high triglyceride concentrations. Electrophoresis studies demonstrated that these individuals had a mutation on the ApoA-I gene due to the substitution of a single amino acid in protein ApoA-I. This protein has 273 amino acids and the Milano mutation consists in the substitution of Arg for Cis in position 173. The subjects with this HDL mutation have a very low rate of both clinical and subclinical atherosclerosis.70 The low rate of cardiovascular events among these subjects inspired the theory that this mutant HDL, containing ApoAIMilano, is much more functional than ApoA-Inative.70 The mutant protein ApoA-IMilano was cloned and bonded to phospholipid palmitoyl-2-oleoyl phosphatidylcholine (POPC) creating synthetic HDL (recombinant HDL [apoA -IMilano]). This type of synthetic HDL has been studied by different research groups in several experimental models with very consistent results regarding the regression of atheromatous plaque.71-77 In addition to multiple preclinical studies, a pilot study in patients with acute coronary syndrome was conducted, and that corroborated the acute regression of coronary atheroma.66
Thus, there is indisputable evidence that the exogenous infusion of HDLMilano is capable of decreasing plaque volume in animal models and in humans. As described, the clinical manifestations of atherothrombosis are dictated to a great extent by the composition of the plaque (ie, vulnerability), and not by the presence of more or less stenotic atheroma. Recently, we published an experimental animal study which analyzed the effect of an acute infusion of HDLMilano on the volume and composition of atheromatous plaques.78 This confirmed the current results obtained by other independent groups,79 since the acute administration of HDLMilano led to a very significant regression in the volume of atheromatous plaque (Figure 5). This effect was evaluated by serial magnetic resonance imaging, as well as by multislice computerized tomography, and it was demonstrated that this imaging technique is also capable of quantifying the changes in atherosclerotic burden,80,and therefore would be a very promising tool for the non-invasive evaluation of antiatherogenic treatments.81 It is striking that plaque regression was accompanied by a qualitative change, as this indicates acute stabilization of atheroma plaque.78 Up to the present, this is the first study that has correlated the regression of atheroma plaque in vivo with changes in plaque vulnerability after the administration of HDLMilano. The mechanism of action involved in such acute beneficial effects seems to be a very strong increase in RTC and an overall antiinflammatory effect that promotes "rescue" from global inflammation identified in the atherothrombotic disease.82 It is worth highlighting the fact that all these beneficial effects on plaque volume and stability were observed after only 2 infusions of HDLMilano (during 4 days). Currently, there are several interventions that have proven effective in stabilizing atheromatous plaque83; however, the time required to induce this effect was relatively long (several months). It is important to emphasize that most recurrent events occur in the acute phase after the first ischemic event.19 Our results indicate that a loading dose of intravenous HDLMilano can induce rapid plaque regression, with a stabilizing effect on active and high-risk plaques.

Figure 5. Atheromatous plaque regression after acute treatment with HDLMilano. Magnetic resonance imaging of cross-sectional studies of the abdominal aorta in atherosclerotic rabbits before and after treatment with HDLMilano or placebo. HDLMilano led to a very significant regression in plaque volume, whereas placebo had no effect. It is important to emphasize that this plaque regression was seen after only 2 infusions of HDLMilano (75 mg of ApoAIMilano/kg). Besides a sharp regression in plaque volume, gene and protein expression studies have demonstrated important signs of plaque stabilization. (Reproduced with permission from Ibáñez et al78).
CONCLUSIONS
High serum concentrations of HDL have been correlated with a decreased incidence of atherothrombotic events; thus, the possibility of increasing HDL concentrations using drugs is being investigated in order to fight this epidemic disease. There are different therapeutic targets to obtain a significant increase in circulating HDL. Several groups of drugs have been studied with promising results for long-term treatment. The benefit of increasing HDL concentrations seems to be additional to the benefits derived from chronic treatment with statins. Intravenous infusion of different forms of reconstituted HDL results in very acute plaque regression, and which is associated with plaque stabilization in the case of HDLMilano. Although confirmation by cardiovascular events studies is still pending, therapy aimed at increasing the HDL concentrations seems to be within reach and may prove to be another step forward in the treatment of atherothrombosis.
Correspondence: Dr. J.J. Badimón,
Atherothrombosis Research Unit, The Zena and Michael A. Wiener Cardiovascular Institute, Mount Sinai School of Medicine,
1 Gustave Levy Place, Box 1030, New York, NY 10029, USA
E-mail: juan.badimon@mssm.edu