A 57-year-old man with spinal arthropathy and treated bilateral carpal tunnel syndrome consulted for anorexia, weight loss, and occasional episodes of epigastric pain, nausea, and vomiting. Over the last year he had experienced dyspnea on exertion. The physical examination was unremarkable.
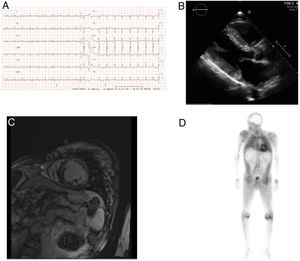
The electrocardiogram showed low voltages, and the echocardiogram depicted severe, asymmetrical left ventricular hypertrophy with septal predominance (19mm); the left ventricular cavity size was normal, with preserved systolic function (left ventricular ejection fraction, 54%) and a pseudonormal diastolic filling pattern (Figure 1A-B).

A, electrocardiogram showing low-voltage in the limbs. B, echocardiogram depicts left ventricular hypertrophy. C, cardiac magnetic resonance shows extensive subendocardial delayed enhancement. D, 99mTc-3,3-diphosphono-1,2-propanedicarboxylic acid scintigraphy depicts myocardial uptake greater than in bone.
On analytic study, performed to investigate a suspected deposit disease, blood count and biochemical findings were normal, with no proteinuria. The serum electrophoretic pattern was normal and free kappa and lambda chains tested negative. Gadolinium cardiac magnetic resonance imaging showed large areas of subendocardial enhancement in both ventricles and the left atrium, consistent with amyloidosis (Figure 1C). On scintigraphy with 99mTc-3,3-diphosphono-1,2-propanedicarboxylic acid, radiotracer deposit in the left ventricle was greater than bone intensity (Figure 1D). Amyloid was not detected in subcutaneous fat or rectal biopsies. The neurological evaluation and sensory-motor nerve conduction studies showed no abnormalities, except for bilateral median nerve involvement.
Five months later, the patient was admitted to the emergency room with acute pulmonary edema. On electrocardiography, the non-dilated left ventricle showed moderate dysfunction and (left ventricular ejection fraction, 38%) and diastolic function had a restrictive pattern. The N-terminal pro-brain natriuretic peptide fraction was 14,879 pg/mL.
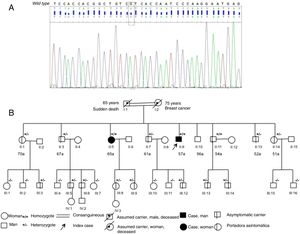
Genetic study, carried out to determine whether the amyloidosis resulted from wild-type transthyretin deposit or a hereditary form of the disease, found a homozygous mutation, Val142Ile (classically, Val122Ile), in the transthyretin gene (Figure 2A). In the family evaluation (Figure 2B) there were no known black ancestors, and the deceased parents were cousins. The mother had no known cardiologic history, but the father had an unspecified cardiac condition since the age of 40 years and died suddenly at 65 years. On study of 7 of the 8 siblings, 2 homozygotes were found and all siblings were asymptomatic. Electrocardiography and echocardiography findings were normal in all, except for 1 sister who had the mutation in homozygosis and showed the following: normal electrocardiography findings, bilateral carpal tunnel syndrome, moderate hypertrophy on echocardiography (septum, 14mm), and left ventricular uptake similar to bone intensity on scintigraphy. Among the nieces and nephews, there were 8 asymptomatic carriers with no electrocardiographic or echocardiographic abnormalities.

A diagnosis of hereditary cardiac amyloidosis due to a mutation in the transthyretin gene was established, and the patient was referred to a reference center for liver and cardiac transplantation, which was carried out successfully.
Transthyretin gene mutations, the most common cause of amyloidosis, lead to neuropathy and often, cardiac disease. Several causal mutations resulting in different phenotypes have been identified.1
The mutation found (Val122Ile) produces cardiac amyloidosis in persons older than 60 years with a phenotype similar to that of wild-type transthyretin amyloid, occasionally associated with carpel tunnel syndrome. Between 3% and 4% of black individuals in the United States are heterozygous carriers of this mutation,2 which is rare in the white population. Although it is considered a rather indolent mutation with late-onset cardiomyopathy, several studies have associated it with greater morbidity and mortality than the wild-type form.3 When the mutation is homozygous, the risk increases, and heart failure develops earlier and is more severe.4
Our patient showed a severe phenotype with rapid progression to heart failure, New York Heart Association functional class II/IV, left ventricular dysfunction, and considerable elevation of N-terminal pro-brain natriuretic peptide.
Genetic study was not only useful for the diagnosis of hereditary amyloidosis, it also helped to understand the rapid course of the condition, as the mutation was homozygous.
New treatments that act at several levels have been developed to detain or delay transthyretin amyloid deposit.5 Some have proven to be effective in randomized clinical trials and have been approved by regulatory agencies. Several of these authorized drugs act by inhibiting hepatic expression of transthyretin with interfering ribonucleic acid (patisiran) or antisense oligonucleotides (inotersen). Other drugs act by stabilizing the transthyretin molecule and preventing its dissociation and deposition. This group includes tafamidis, which has been proven to reduce cardiovascular mortality and hospital admissions.6 Other stabilizers are under development. Finally, it may be possible to eliminate amyloid deposits by antibodies directed toward transthyretin or by molecules such as doxycycline. Several trials are currently underway to evaluate these compounds.5
Genetic screening enabled identification of 15 carriers who require close monitoring and may benefit from the early start of these new treatments to slow the development of the disease and improve the prognosis.
An early diagnosis in our patient would have allowed initiation of effective drug treatment and avoided progression to the terminal phase requiring transplantation. Fortunately, the carriers identified, particularly the sister in an early phase of the disease, will benefit from the available treatments.