Keywords
INTRODUCTION
Compared to the left ventricle (LV), until recentlythe right ventricle (RV) has received very littleattention in patients with acquired heart disease and only slightly more in those with congenital heartdisease (CHD). Whether RV dysfunction affectsprognosis in acquired heart disease is largely unclear.In patients with CHD, however, emerging data islinking RV dysfunction and fibrosis to quality of life,symptoms and prognosis. The RV in CHD patientsis often, but not always, the subpulmonary ventricle,as for example in patients with transposition ofthe great arteries, where it supports the systemiccirculation. As a consequence, RV function maybe impaired either as a result of pressure or volumeoverload or a combination of the two. Accurateunderstanding of RV anatomy and assessment ofRV volumes, and function, is thus paramount forclinicians involved in the care of CHD patients. Thisarticle provides an overview of RV anatomy, andreviews the central role of the RV in CHD.
ANATOMY
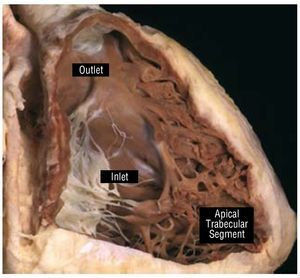
The RV is composed of 3 different segments: inlet,apical trabecular, and outlet (Figure 1). The inletcomponent extends from the tricuspid valve (TV)annulus to the insertion of the papillary muscles in the ventricular wall and thus surrounds and supportsthe leaflets and tension apparatus of the TV. TheTV has 3 leaflets: the septal, antero-superior, andinferior (or mural). Its most characteristic featureis the presence of tendinous cords attaching itsseptal leaflet to the ventricular septum, which,along with, the apical displacement of the TV, canbe used to distinguish it from the mitral valve andthe morphological LV during echocardiography. Another useful marker for identifying themorphological RV is the moderator band, a broadmuscular strap that crosses the ventricular cavity.

Figure 1. This window dissection shows the 3 components of a normalright ventricle. Courtesy of Professor Y. Ho.
The apical trabecular component involves theRV body and apex and has characteristically coarsetrabeculations. The outlet portion is muscularand elongated, extending to the pulmonary valve,which does not have a real valvar annulus. Thismore triangular shape of the morphological RV,distinguishes it from the more conical morphological LV.
The 2 ventricles also differ significantly in myofiber orientation. While the RV myofibersare circumferential and longitudinal, the LVwall contains obliquely arranged myofibers onthe surface, and longitudinal myofibers in thesubendocardium, with circumferential myofibresin between, in a sandwich fashion.1 The free wallof the normal RV is usually 3 to 5 mm in thickness,but in conditions of pressure overload the RV wallthickness may even exceed that of the LV, as doesits myoarchitecture.2
THE PRESSURE LOADED RIGHT VENTRICLE
The 2 most common models of pressure loadedRV are RV outflow tract (RVOT) obstruction andRV as a systemic ventricle.
RIGHT VENTRICULAR OUTFLOW TRACTOBSTRUCTION
Congenital obstruction of RVOT is mostcommonly due to pulmonary valve stenosis (PS), butmay also be the result of abnormalities at the mid-RV, the infundibulum, the supravalvular region, orthe branch/peripheral pulmonary arteries3 (Table).

Pulmonary Valve Stenosis
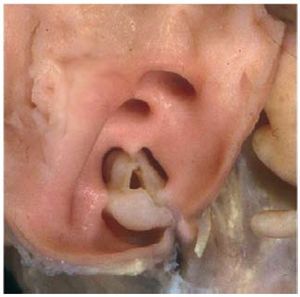
Isolated PS is found in 80%-90% of all patientswith RVOT obstruction, and is almost alwayscongenital. It is possible to identify 3 morphologicaltypes of stenotic pulmonary valve: a) dome-shaped; b) dysplastic; and c) bicuspid or unicuspid.4 Thelatter type is uncommon. The classic form of PS is thedome-shaped pulmonary valve, which may have 2-4raphes, but there is no separation into valve cusps5 (Figure 2). It is characterized by a mobile valve withnarrow central opening.6 Its familial inheritance islow, varying between 1.7%-3.6%.7,8

Figure 2. Dome-shaped pulmonary valve viewed from the arterial aspect.Courtesy of Professor Y. Ho.
A dysplastic pulmonary valve is present in 10%to 20% of patients with PS.9,10 Dysplastic valves aretrileaflet with markedly thickened leaflets composedof disorganized myxomatous tissue and little, if any,fusion. This is usually associated with a hypoplasticventriculo-arterial junction. This entity is the most common finding in patients with Noonan syndromeand valvar PS.10
PS, when significant, results in compensatory RVhypertrophy, especially at the infundibular level.When prominent, RVOT hypertrophy can lead tosecondary dynamic subvalvar stenosis. PS can alsoresult in post-stenotic dilatation of the pulmonarytrunk, which is common in the doming form of PSand often extends to the proximal left pulmonaryartery. This is thought to be the result of the high-velocity jet through the narrow valve orifice,which is anatomically aimed more toward the leftpulmonary artery (natural continuation of themain pulmonary artery), and can produce unequaldistribution of blood flow in favor of the left lung.11 However, intrinsic abnormalities of the pulmonaryarterial wall also contribute to the pulmonary arterydilatation. Interestingly, post-stenotic dilatationof the pulmonary artery is rare in patients withdysplastic pulmonary valves.
A hypertrophied RV can maintain its function foryears, even when RV pressures are near systemic.It is, in fact, an exaggeration that the RV dilatesand fails early in life when chronically exposed tohigh pressures; indeed, as long as sinus rhythmis preserved, and there is no additional volumeoverload, the RV is usually able to maintain itsfunction well into the 4th or 5th decade of life.12
Most patients with PS remain asymptomatic formany years, even when stenosis progresses frommoderate to severe. Therefore, diagnosis is notuncommonly made in adulthood. Both in infancyand adulthood, this condition is usually detectedwhen a characteristic murmur is heard on physicalexamination, and is thereafter confirmed bytransthoracic echocardiography. Symptoms usuallydevelop when RV pressure exceeds 50% of systemicpressure, limiting RV output and pulmonaryperfusion. Typical symptoms include exertionaldyspnea and fatigue. Chest pain, syncope or evensudden cardiac death may also occur in patients withsevere PS, which is thought to result from decreasedmyocardial perfusion caused by inadequate cardiacoutput during exercise, leading to ischemia andventricular arrhythmias.3
The auscultatory findings in PS are quite distinct;the mobile pulmonary valve creates an ejection clickthat decreases with inspiration. The more severe thestenosis, the earlier in systole the click occurs, untilit merges with the first heart sound and becomesinaudible. There is also a crescendo-decrescendoejection systolic murmur, maximal at the upper leftsternal border. Pulmonary regurgitation (PR) isuncommon in this setting. A 4th heart sound is oftenheard at the lower left sternal border in patients withsevere stenosis. When a 3rd heart sound is heard, thepresence of an atrial septal defect (ASD) should besuspected.
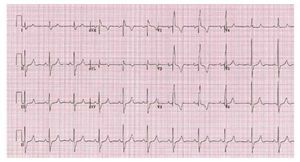
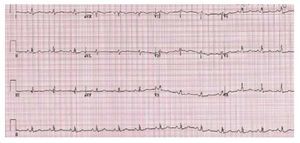
The electrocardiogram is usually normal when theRV systolic pressure is <60 mmhg as the lesion severity worsens electrocardiographic evidence of right atrial enlargement axis deviation and rv hypertrophy may appear 13 In severe PS a pure R,Rs, or QR is the usual pattern in the right precordialleads, and the R wave is usually greater than 20 mm(Figure 3).

Figure 3. Patient with severe pulmonarystenosis. The ECG shows right bundlebranch block with right ventricularhypertrophy.
The chest radiograph often has distinctivefeatures, with normal heart size and a dilated centralpulmonary artery. This sign can be absent in patientswith dysplastic valve. Vascular fullness in the left lungbase greater than the right (Chen's sing) is due to thepreferential flow. In severe PS pulmonary vascularmarkings can be diminished.11 The right atrium andRV can be dilated if there is RV decompensation orwhen PS is associated with an ASD.
Echocardiography is the diagnostic method ofchoice. Continuous wave Doppler is useful forobtaining peak instantaneous gradient through theRVOT/pulmonary valve. However, distinguishingbetween valvar stenosis and subvalvular dynamicstenosis secondary to infundibular hypertrophycan become challenging. Subvalvular dynamicobstruction, in fact, often accompanies severevalvular PS and is characterized by a late-peaking jetsimilar to that of dynamic LV outflow obstruction.Color and pulsed wave Doppler can also be usedto locate the site of obstruction and, therefore,distinguish between valvular, subvalvar (RVOT) orsupravalvar PS. Echocardiography can also provideinformation on valve mobility, RV size and function,as well as the presence of poststenotic dilatation.Cardiac magnetic resonance (CMR) can provideadditional information for assessing PS and locatingthe exact area of obstruction. In addition, CMR is the gold standard method for quantifying RV sizeand function.
The first operation for PS was performed in 1948and consisted of pulmonary valve commissurotomy.Subsequent procedures in the early 1950s involvedclosed valvotomy with transcatheter methods(Brock procedure). Later, open valvotomy yieldedsignificantly better results. If the valve is dysplastic,partial or total valvotomy may be required; ifthere is annular or pulmonary trunk hypoplasia,a transannular patch may become necessary. Allof these procedures invariably result in variousdegrees of PR, which is well tolerated for manyyears. Initially, the RV compensates by dilatation,maintaining contractility and stroke volume.Eventually RV dysfunction ensues and patientsdevelop symptoms, such as dyspnea, fatigue andsubstantial arrhythmia.
The long-term outcome of patients with PS isexcellent. In the Second Natural History Study ofCongenital Heart Defects, there was no significantdisease progression in patients with a peak gradient <20 mm hg while there was a 20 chance of requiring repair in those with peak gradient between 25 and 49 the majority patients gradients 50 mm Hg requiredintervention. 14 Studies of patients who receivedsurgical valvotomy in childhood have shown thatoutcome is excellent in this cohort, with a mortalityrate <5 after 25 years of follow-up 14 However,one-third of the patients will develop significant PRand will require re-intervention in later years (re-intervention rate 9%-40%; mean time to pulmonaryvalve replacement [PVR], 33 years). 15-17 Atrial or ventricular arrhythmia, exercise intoleranceor cardiomegaly on chest x-ray in patients with previous prior surgical pulmonary valvotomyshould raise the suspicion of PR. Subjectivemeasures of exercise intolerance may be unreliablein these patients due to the chronic nature ofPR and the slow progression of the disease.Cardiopulmonary exercise testing is, thus, essentialfor objectively quantifying exercise intolerance andmonitoring change over time. Moreover, serialechocardiography is important for detecting RVdysfunction and the development of significanttricuspid regurgitation (TR), which should promptreferral for PVR. 12
Since percutaneous balloon valvotomy wasintroduced in 1982,18 it has become the treatmentof choice for patients with classic domed valvularPS. The American College of Cardiology/AmericanHeart Association guidelines recommend balloonvalvotomy for asymptomatic patients with domed-shaped pulmonary valve and a peak instantaneousDoppler gradient >60 mm Hg or a mean Dopplergradient >40 mm Hg. For symptomatic patients,balloon valvotomy is indicated when a peakinstantaneous Doppler gradient >50 mm Hg or amean Doppler gradient >30 mm Hg is present.19Long term outcome after balloon valvotomyis excellent,20 with a low rate of restenosis. Thelatter was found to be more common when aresidual gradient was present immediately afterthe procedure.21 Significant PR is encountered in5% of patients after balloon valvotomy. In thiscohort, a pulmonary regurgitant fraction >15% was associated with a lower peak VO2.22 Whether balloon valvotomy will be associated with similarrates of re-intervention for PR in the long termremains speculative.
Double-Chambered Right Ventricle
Double-chambered RV (DCRV) is usuallyassociated with a ventricular septal defect (VSD).It is characterized by aberrant hypertrophiedmuscular bands that divide the RV cavity into aproximal high-pressure and a distal low-pressurechamber (Figure 4). This is in contrast to RVOTobstruction in tetralogy of Fallot (ToF), wherethe main level of obstruction is usually at theinfundibular level.

Figure 4. Patient with double-chamberedright ventricle. The arrow shows thehypertrophied muscle band.
The severity of the DCRV obstruction tends toincrease with time and may only become manifestduring adulthood. Clinically, patients with DCRVand intact ventricular septum resemble patients withisolated PS. When a VSD is present, the clinicalfeatures may be dominated by the VSD rather thanthe DCRV, early in the course of the disease.
On examination, a loud pansystolic crescendodecrescendo murmur is typically present,often accompanied by a thrill, which may beundistinguishable from that of isolated PS, but forthe absence of the ejection click. A murmur relatingto the associated VSD may be present, unlessthe VSD opens into a high-pressure (proximal)chamber.
The electrocardiogram typically demonstratesRV hypertrophy, which is not what is expectedin cases of isolated restrictive VSD. Two-dimensional echocardiography is usuallydiagnostic, identifying the degree and locationof the obstruction and the presence of a VSD.However, the VSD may be difficult to visualizewhen it opens into the high-pressure chamber.CMR provides complementary information onthe anatomy and physiology.
The indications for surgery in DCRV are similarto those for valvar PS.19 Muscular resection andoutflow-enlarging procedures have been veryeffective, with excellent long-term results and verylow rates of recurrence.23
Pulmonary Stenosis and Pregnancy
When RV function is preserved, isolatedRVOT obstruction is usually well toleratedduring pregnancy, even when the lesion is severe.Nevertheless, when women with severe PS developrecurrent atrial arrhythmia and/or early right heartfailure during pregnancy, balloon valvotomy shouldbe considered.
SYSTEMIC RIGHT VENTRICLE
A morphological RV in the systemic positionin adulthood is most commonly encountered inpatients with congenitally corrected transposition(ccTGA) and those with transposition of the greatarteries (TGA) following atrial switch procedures(Mustard or Senning). In the systemic position,the RV changes its myofiber architecture toresemble the "sandwich" pattern encountered inthe normal LV.4 Moreover, in the systemic RVthere is predominant circumferential rather thanlongitudinal free wall shortening.5 These changesallow the RV to adapt and function in the systemicposition to a large extent and for a number ofdecades.
Congenitally Corrected Transposition
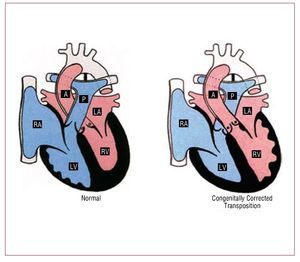
ccTGA is a rare condition, accounting for <1 of all chd it consists atrioventricular av and ventriculoarterial va discordance: the right atrium is connected to morphological lv which gives rise pulmonary artery whereas left rv aorta also generally but not universally anterior great arteries may be side by rather than crossing figure 5 situs abnormalities are common in this entity fact cctga one cardiac defects most commonly associated with dextrocardia should suspected when latter abdominal solitus additional lesions present 95 cases include ebstein-like anomaly tv 90 vsd 70 ps 40 complete heart block 2 risk per year 24

Figure 5. Normal heart and congenitallycorrected transposition. A indicatesaorta; LA, left atrium; LV, left ventricle;P, pulmonary artery; RA, right atrium; RV,right ventricle. Courtesy of Professor Y.Ho.
Patients with ccTGA typically remainasymptomatic until the 3rd or 4th decade of life butmay become symptomatic earlier when additionalhaemodynamic lesions are present.25 The most common symptoms include dyspnea on exertionand palpitations or syncope secondary to atrialarrhythmias or complete heart block. Patients withVSD and PS may present with progressive cyanosis.
Echocardiography is the diagnostic method ofchoice. The apical four-chamber and subcostalviews are most helpful in determining the situs andAV-VA connections. Ventricular morphologyis best assessed by examining the AV valves: theTV is always closer to the apex compared to themitral valve and its septal leaflet has chordalattachments to the inlet septum. The relation ofthe two ventricles and the great arteries are moreside by side than the usual anteroposterior. It isimportant to identify associated lesions such asVSD, which is usually perimembranous, as wellas the presence of LV outflow tract obstruction,which may be due to either an aneurysm of themembranous septum, a fibrous membrane, ormobile subpulmonary tissue "tags." Moreover,accurate assessment of systemic RV function andthe degree of systemic AV valve regurgitation isalso fundamental in the assessment of ccTGA.CMR provides complementary information onthe anatomy and a more accurate estimate of RVsize and function.
The prognosis of ccTGA greatly depends onthe presence and severity of associated lesions.In the absence of the above, patients with ccTGAmay survive until the 7th or 8th decade of life.26 However, the incidence of systemic ventriculardysfunction and congestive heart failure increaseswith age, even in the absence of associated lesions(more than 1 in 3 will develop congestive heartfailure by the 5th decade of life).27 The presenceof significant TR and/or RV dysfunction isassociated with a significantly higher mortality27,28 and a higher risk of developing decompensatedheart failure. The rate of deterioration of systemic RV function in the presence of significant TR is muchmore rapid than that of systemic LV in the presenceof mitral regurgitation. The factors responsible forthis remain unclear. Ventricular geometry is likelyto play a role in this, as RV dilatation is more likelyto cause severe annular dilatation and rapidlyaggravates TR and in turn RV dysfunction.29 In addition, myocardial perfusion may not be adequate to sustain the workload of a severelyhypertrophied systemic RV, making it vulnerableto perfusion mismatch and ischemia.30 When systemic RV function deteriorates in the presence ofsystemic AV valve regurgitation, particularly whenintrinsic abnormalities of the valve are present, TVreplacement should always be considered, beforeirreversible systemic RV dysfunction ensues.19
While there is evidence of neurohormonalactivation in patients with ccTGA, little is knownabout the efficacy of heart failure medication, suchas angiotensin converting enzyme inhibitors (ACEI)and beta-blockers.31 Data are conflicting and manystudies combine patients with ccTGA and Mustardor Senning procedures, the latter may responddifferently to ACEI due to abnormal AV function.A small pilot study on patients with ccTGA orSenning procedure has suggested that carvedilolmay improve RV function. However, beta-blockersshould be used with caution due to the propensityfor conduction abnormalities and complete AVblock,32 unless patients are paced.
Complete Transposition of the Great Arteries
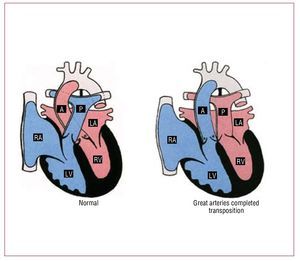
Complete TGA accounts for approximately 5% ofall newborns with a congenital cardiac malformation.In these patients, the aorta arises from themorphological RV, and the pulmonary artery from the morphological LV (Figure 6). The great vesselsare parallel, with the aorta anterior and, usually, tothe right of the pulmonary artery. Associated lesionsare present in one-third of patients.

Figure 6. Normal heart and transpositionof the great arteries. A indicates aorta; LA,left atrium; LV, left ventricle; P, pulmonaryartery; RA, right atrium; RV, right ventricle.Courtesy of Professor Y. Ho.
Nowadays, the treatment of choice for patientswith TGA is the arterial switch procedure, which has been applied for more than two decades.However, most of the adults with TGA alive todayhave had an atrial switch procedure (Mustardor Senning operation) which results in the RVsupporting the systemic circulation. These patientsmay develop systemic ventricular dysfunction,arrhythmias or baffle-related problems, andwarrant annual follow-up in specialized adultcongenital heart disease (ACHD) centers.19 In thesepatients, RV function may gradually deteriorateand this may be associated with aggravating TR.Only patients with significant TR due to primaryTV disease (which may be iatrogenic from VSDclosure or due to other factors such as endocarditis)may benefit from TV replacement.19 Assessmentof RV function in these patients is paramount,but also challenging. Although transthoracic echoassessment of adults is limited to semiquantitativedata in this setting, key hemodynamic informationon RV may be obtained by experienced hands.Systemic ventricular free wall long-axis excursionhas been shown to predict exercise capacity bythese patients.33 Echocardiography also provides information on baffle patency, baffle leaks andvalvar regurgitation.
CMR is considered the gold standard for thestudy of RV size and function in this population.Moreover, the systemic and pulmonary venouspathways can be evaluated more accurately. WhenCMR is contraindicated, gated CT-angiographymay be an alternative.
Both bradyarrhythmias and tachyarrythmias arecommon long after atrial switch for TGA. Sinusnode dysfunction is frequent and 11% of patientswill need pacemaker implantation at some point.34 Almost a quarter of patients develop atrial flutteror fibrillation 23 years after the atrial switchprocedure.35 The extensive atrial scarring followingatrial surgery creates the perfect substrate for atrialarrhythmias, which may become life threateningwhen fast or in the presence of significant pathwayobstruction. Antiarrhythmic drugs with negativeinotropic effect are rarely used when systemic RVdysfunction is present. Radiofrequency ablationfor atrial arrhythmias can be challenging, but hasa success rate of approximately 70% in experiencedhands.36
Atrial pathway obstruction and leaks, whileinfrequent, should always be excluded in thepresence of symptoms. Superior vena cava (SVC)obstruction is more common and may cause "SVC syndrome," although this is uncommon becausethe azygos system usually compensates for it.Patency of the superior systemic venous pathwayshould always be assessed before implantation of atransvenous pacing system. Small baffle leaks arerelatively more common. They are usually smalland haemodynamically insignificant but may allowparadoxical emboli and cause cyanosis at rest orduring exercise. When the LV is enlarged, contrastechocardiography with agitated saline should beperformed to rule out a significant pathway leakwith predominant systemic to pulmonary shunt(equivalent of RV enlargement in concordantheart with an ASD).
Pulmonary arterial hypertension occurs in about7% of patients after atrial switch procedures forTGA.37,38 The pathogenesis is unknown, but risksfactors include operation at >2 years of age39 and shunting at the ventricular or great artery level priorto repair.40 Pulmonary venous pathway obstructionshould always be investigated as a cause of pulmonaryhypertension in this setting.
Systemic Right Ventricle and Sudden CardiacDeath
The majority of patients with systemic RV diesuddenly.41,42 Age, systemic ventricular dysfunction, New York Heart Association functional class,43 supraventricular arrhythmia35 and QT dispersion44have all been associated. Recently, Schwerzmannet al43 reported that a QRS duration ¡Ý140 ms isassociated with lower functional class, worse systemicRV function and a higher mortality. Automatedimplanted cardioverter defibrillators (AICD) havebeen utilized recently in this setting, although theirrole remains unknown.
Systemic Ventricle and Pregnancy
The risk of pregnancy in patients with systemicRV depends on systemic ventricular function,the presence of significant hemodynamic lesionsand functional capacity.45 Pregnancy may causedeterioration in systemic RV function, even thoughthe long-term impact of pregnancy on systemic RVfunction remains unclear. A mortality rate of up to4% has been reported in a small series of womenwith systemic RV, and this should be discussedduring preconception counseling.46 Patients withgood or mildly impaired RV systolic function andno pathway obstruction are at low risk.47,48 During pregnancy, clinical assessment should focus onearly signs of heart failure and arrhythmias. If atrialtachycardia occurs, direct current cardioversioncan be performed safely to restore sinus rhythm.Aspirin throughout pregnancy should beconsidered in patients with previous history ofatrial arrhythmias.
THE VOLUME LOADED RIGHT VENTRICLE
The 3 most common lesions associated with RVvolume overload are ASD, pulmonary regurgitationin the setting of ToF and TR in the setting of Ebsteinanomaly. In this review, we will focus on the latter2 conditions.
TETRALOGY OF FALLOT
ToF is the most common form of cyanotic CHDafter the first year of life. It consists of a largemalaligned subaortic VSD, the aorta riding up overthe septal defect (when more than 50% patientsfall into double outlet RV subgroup), RVOTobstruction and RV hypertrophy. Pulmonarybranch stenosis or pulmonary branch hypoplasiamay be present, with some patients presentingwith pulmonary atresia. Associated anomaliesinclude ASD, AV septal defect (more common inpatients with Down syndrome) and a right-sidedaortic arch, which can be found in up to 25% ofpatients. Coronary artery anomalies may alsobe present, commonly involving the left anteriordescending coronary artery, which arises from the right coronary sinus and crosses the RVOT (3% ofcases).49
Most patients with ToF, nowadays, undergototal repair early in life. This involves closure ofthe VSD and relief of the RVOT obstruction. Thelatter may require RVOT/transannular patch,which disrupts the integrity of the pulmonary valve "annulus," or pulmonary valvotomy/valvectomy ifthe pulmonary valve is abnormal. An extracardiacconduit between the RVOT and main pulmonaryartery may be necessary in the presence ofpulmonary atresia or an anomalous left coronaryartery crossing the RVOT.
The most common sequela after RVOT patchaugmentation is significant PR. Severe chronicPR may lead to RV dilatation and RV systolicdysfunction,50 with a propensity to clinicalarrhythmia and sudden cardiac death.51,52 It is known that after 2 decades of exposure to significantvolume overload, RV systolic function deteriorates,resulting in progressive exercise intolerance and ahigh risk of arrhythmias, both supraventricular andventricular. Patients should, thus, be consideredfor PVR when significant RV dilatation is present,before the onset of irreversible RV dysfunction.Reliance on symptoms only for deciding the timingof PVR may be misleading, as patients often becomesymptomatic when RV function becomes severelyimpaired. Objective assessment of exercise capacitymay be more reliable, and serial exercise testing mayhelp identify changes in exercise capacity, which maynot be perceived by the patient. Surgical interventionis also indicated when patients with severe PR andenlarged RVs develop moderate to severe TR, orsymptomatic atrial or ventricular arrhythmias.19 Indeed, no single parameter, but several variables,should be considered when deciding the correcttiming of PVR.
Echocardiography remains the most widelyemployed imaging modality for assessing patientswith ToF. However, CMR is the gold standard forboth PR quantification and RV volumetric analysis.Patients with repaired ToF, especially those withprogressive TR or other sequelae, should have anechocardiogram annually and a CMR every 2-3years.19 Quantification of PR with echocardiographycan be challenging for inexperienced operators.Early equalization of RV and PA pressures can be asign of significant PR in the absence of a restrictiveRV. A pressure half-time <100 ms 53 and a PR index(ratio between PR duration and total diastole) lowerthan 0.77 has 100% sensitivity and 85% specificityfor identifying patients with pulmonary regurgitantfraction greater than 25% in CMR 54 (Figure 7). RVsize and function should also be evaluated.

Figure 7. Patient with repaired tetralogy ofFallot and severe pulmonary regurgitation.Pulmonary regurgitation index is 58%(b/a).
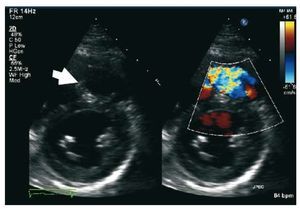
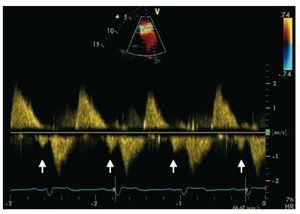
Pulsed Doppler late-diastolic forward pulmonaryflow, coincident with atrial systole, present throughout respiration, and associated withretrograde vena caval flow, defines the so-called "restrictive RV physiology" in this setting (Figure 8). This physiology is commonly present early afterToF repair, and has been shown to be associatedwith low cardiac output, leading to a longerintensive care stay.55,56 However, there are data tosuggest that restrictive RV physiology late afterrepair of ToF is associated with smaller RV size,shorter QRS duration and better exercise capacity,possibly by counteracting the effects of PR and byalso contributing to forward pulmonary flow andthus to cardiac output.57,58

Figure 8. Patient with repaired tetralogyof Fallot and restrictive right ventricle. Thearrows show the late diastolic forwardflow ("a" wave).
The unusual shape of the RV cavity and theunpredictable manner in which it dilates makeaccurate quantitative analysis by echocardiographicor radionuclide angiographic techniques difficult.CMR permits more accurate and reproduciblequantitative analysis of RV dimensions andfunction in experienced hands. The optimal "cut-off" value beyond which PVR should berecommended remains controversial. Therrien etal59 reported that RV volumes do not normalizein patients with a preoperative RV end-diastolicvolume >170 mL/m2 and RV end-systolic volume >85 mL/m2. Oosterhof et al60 reported that thereis no threshold above which RV volume does notdecrease after surgery, but normalization of RVvolumes is only achievable when RV end-diastolicvolume does not exceed 160 mL/m2 or RV endsystolic volume 82 mL/m2. Early PVR in patientswith RV end-diastolic indexed volume of 150 mL/m2 was recently shown to lead to normalization ofRV dimensions61 and is considered by many as a cut-off value, although other parameters need to betaken into account.
Most patients after ToF repair have right bundlebranch block as well as signs of RV hypertrophyon the electrocardiogram. Prolongation of QRSduration correlates well with progressive RVdilatation and dysfunction. A mechano-electricalinteraction has been postulated, reflecting delayedconduction and increased likelihood of malignantventricular arrhythmias.52 Therefore, followingQRS prolongation over time is important, witha QRS rate of change ¡Ý4ms/year associatedwith an increased risk of sustained ventriculartachycardia and sudden cardiac death.51 Other predictors of ventricular tachycardia and suddencardiac death include a QRS duration ¡Ý180 msand LV dysfunction,51,52 even though the latter has a low predictive positive value when takenalone (29% vs 93% when considered together withQRS duration).62 Stratification for sudden cardiacdeath remains challenging for individual patientswith ToF, and the role of primary prevention withAICD implantation in high risk patients (largeRVOT aneurysms,63 QRS>180 ms,52 inducibleventricular tachycardia,64 extensive late gadoliniumenhancement65 or LV dysfunction62) remainsuncertain.66 Atrial arrhythmias and reducedheart rate variability has also been shown to be amarker of increased morbidity and mortality in thispopulation.
Although pregnancy is well tolerated in patientswith regurgitant lesions, severe PR after ToFrepair has been reported as a maternal risk factorduring pregnancy.67 Furthermore, the risk for adverse maternal events associated with pregnancyincreases if RV or LV dysfunction or hypoplasticpulmonary arteries are present. The latter, however,is uncommon. Pregnancy is a relatively low-riskendeavor for most women with repaired ToF and,during pregnancy, periodic assessment should focuson signs of right and left heart dysfunction andclinical arrhythmia.68
EBSTEIN ANOMALY
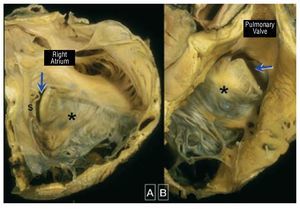
Ebstein anomaly is a rare and complexdisorder accounting for <1 of all cases chd it is characterized by adherence the septal and posterior tv leaflets to underlying myocardium due failure delamination during embryonic development this leads apical displacement functional annulus leaflet more than anterior may be severely deformed forms a large sail-like intracavitary curtain which can even lead rvot obstruction figure 9 inlet portion rv functionally integrated with right atrium atrialized become disproportionately dilated varying degrees wall thinning 69 These anatomical and functionalabnormalities may cause significant TR, whichresults in right atrial and ventricular dilatation.The LV is typically small ("compressed") and isoften intrinsically abnormal in its function.

Figure 9. Ebstein malformation of thetricuspid valve viewed from the rightatrium (A) and from the right ventricularoutflow tract (B). In this heart the sail-likeleaflet (*) has a limited free margin. Thisresults in a key-hole aperture (arrow)between the anterior leaflet and theunderdeveloped septal leaflet (s) thatis the effective valvar orifice displacedtoward the outflow tract. Courtesy ofProfessor Y. Ho.
Ebstein anomaly can be associated with differentcongenital heart defects, such as ASD (present in80%-94% of patients), PS, pulmonary atresia orhypoplastic pulmonary arteries, subaortic stenosis,bicuspid aortic valve, mitral valve prolapse andVSD. In addition, patients with Ebstein anomalyhave higher prevalence of accessory conductionpathways (Wolff-Parkinson-White syndrome).
The first classification of Ebstein anomaly wasdescribed by Carpentier et al70 in 1988. Ebsteinanomaly was classified in 4 categories:
- Type A: the volume of the true RV is adequate.
- Type B: there is a large atrialized component ofthe RV but the anterior leaflet moves freely.
- Type C: the anterior leaflet is severely restricted inits movement and may cause significant obstructionof the RVOT.
- Type D: near-complete atrialization of theRV, with the exception of a small infundibularcomponent.
This classification, though fairly simplistic, maystill be useful in deciding the type of surgery.
The extended Glasgow Outcome Scale71 is a more recent echocardiographic grading score forEbstein anomaly which provides information aboutprognosis. The ratio of the combined area of theright atrium and "atrialised RV" is compared tothat of the functional RV and left heart (grade 1, <0 5 grade 2 0 5-0 99 3 1-1 49 4 ý1 patients in are at higher risk of early mortality a ratio 1 to indicates decreased death with an 10 while the rate childhood goes up 45 <1 indicates 92 survival beyond childhood p
The clinical presentation of Ebstein anomalydepends on the severity of TR, RV function, thesize of the right atrium, right atrial pressure andthe presence of right to left shunting. Althoughmost patients with Ebstein anomaly surviveinfancy, severe heart failure and cyanosis mayoccur early in life. However, such symptomsmay improve as pulmonary vascular resistancedecreases. Exercise intolerance with dyspnea and/or fatigue, symptomatic arrhythmias and right-sided (congestive) heart failure are the mostcommon presenting symptoms or signs duringadulthood. Generally, the younger the age atclinical presentation, the more severe the anatomicand hemodynamic derangement. When an ASD orpatent foramen ovale is present, cyanosis may benoted at rest or during exercise and paradoxicalembolism is possible. Less commonly, left to rightatrial shunting may be present when there is onlymild TR and little rise in right atrial pressure, whichin turn may contribute to RV dilatation.
Tachyarrhythmias are a more common mode ofpresentation of Ebstein anomaly in adults than heartfailure, affecting 20% to 30% of patients.72 These arerelated to right chamber enlargement or the presenceof one or more accessory conduction pathways.
On examination, the jugular venous pressureis rarely elevated in Ebstein anomaly, even inpresence of severe TR, due to the severely enlargedright atrium, which does not allow propagation ofregurgitant flow to the SVC. Peripheral cyanosismay be present, especially when right to left atrialshunting and severe Ebstein anomaly and a lowcardiac output are present. A RV lift is oftenpresent and the pulse volume may be low. The firstheart sound is loud, and there may be a systolicclick relating to the enlarged sail-like anteriorleaflet. A holosystolic murmur, may be heard inthe presence of significant TR and may increase oninspiration, but only when an adequate functionalRV is present.
The electrocardiogram is abnormal in most of thepatients with Ebstein anomaly. A tall and broad Pwave is present as a result of right atrial enlargement.A complete or partial right bundle branch block istypically present. The QRS complex over the right-sided chest leads may be of low voltage (Figure 10). Accessory pathways and first degree AV block mayalso be present.

Figure 10. Patients with Ebstein anomaly.The ECG shows low QRS voltage and tallP waves.
The cardiac silhouette may vary from normalto severely enlarged with globular heart. Thepulmonary vascularity may be normal or decreased.A cardiothoracic ratio >0.65 relates to poorprognosis.
Echocardiography permits accurate evaluation ofthe TV leaflets and the size and function of the rightand left chambers. The main echocardiographic feature of Ebstein anomaly is apical displacementof both, the septal and posterior tricuspid leaflets,exceeding 20 mm or 8 mm/m2 body surface area inadults.73 The site and degree of TR and the feasibilityof TV repair can also be assessed. Attention shouldbe made not to underestimate the severity of TRwhen this is severe ("free"), as this becomes laminar,with low velocity, and an early-peaking Dopplerprofile.12 CMR may supply important additionalinformation in cardiac structure and function,essential in the pre-operative period.
Appropriate timing for TV repair/replacementis important, taking into account predictors ofoutcome such as severity of Ebstein anomaly,RV and LV dysfunction and functional class. Inthe earlier era, a cardiothoracic ratio >65% wasconsidered an indication for surgery. Nowadays,however, it is generally agreed that surgery shouldbe offered before severe cardiomegaly develops,as this is associated with severe RV dilatationand systolic dysfunction. Many would consider acombination of the following as good indications forsurgery: increasing debilitating symptoms or gradualdeterioration in exercise capacity (with or withoutcyanosis), paradoxical embolism, progressivecardiomegaly on chest-x-ray and progressive RVdilatation or deterioration of RV systolic function.19 Although it has not been shown whether TVrepair or replacement provides a better long-termoutcome, TV repair is preferable whenever possible.Concomitant arrhythmia surgery in patients withprevious history of atrial arrhythmias may provideadditional benefit.
Pregnancy is generally well tolerated in noncyanotic women with Ebstein anomaly, with orwithout prior repair, who are asymptomatic orminimally symptomatic. However, an increased riskof pregnancy-related complications and fetal loss has been described amongst such women, particularly inthe presence of cyanosis and/or severe symptomaticTR or arrhythmias.74 These patients should beconsidered for intervention prior to pregnancy. Thereported rate of recurrence of CHD in offspringof patients with Ebstein anomaly is approximately4%.75
CARDIAC RESYNCHRONIZATION THERAPYAND THE RIGHT VENTRICLE
Cardiac resynchronization therapy (CRT)improves haemodynamic parameters and functionalcapacity and reduces morbidity and mortality inpatients with acquired heart failure.76 However,there is little evidence to support the use of CRT inACHD patients. There is some evidence suggestingthat CRT may acutely improve hemodynamics andfacilitate weaning from cardiopulmonary bypassin this setting. Janousek et al77 demonstrated thatCRT improves systemic RV function in patientswith native or LV pacing-induced electromechanicaldelay. However, CRT implantation in ccTGA orTGA patients can be challenging due to the coronarysinus and coronary venous anatomy.78 Moreover,even when feasible, it is difficult to decide whichpatients will benefit from this therapy and clearlymore data are needed in this area.
Biventricular dysynchrony may also be present inpatients with right bundle branch block after ToFrepair and commonly is associated with reducedglobal and regional LV function.79 It is unclear, however, how CRT can be successfully appliedin this setting. Moreover, the presence of areas ofRV late activation in the free wall,80 in portions ofthe interventricular septum80 and in the outflowtract81 probably means that the exact target forRV resynchronization is different from patient to patient. Currently there is not enough evidence torecommend routine CRT in this population andagain more data are required.
CONCLUSION
The RV, with its complex geometry andunique adaptive mechanisms in CHD, remainsa challenge in adult cardiology. Maintaining anadequate RV function and avoiding excessive RVdilatation is essential both in the subpulmonary and systemic position and will influence exercisecapacity as well as short and long term morbilityand mortality. While hemodynamic interventionsto relieve volume overload from TR or PR seemto improve hemodynamics and functional class,timing of valve surgery is evolving. These patientsare best assessed and followed by in tertiarycentres by cardiologists expert in ACHD. Noveltherapies such as AICD and CRT hold promisefor the failing systemic RV, even though furtherstudies are required.
ABBREVIATIONS
CHD: congenital heart disease
LV: left ventricle
PR: pulmonary regurgitation
PS: pulmonary stenosis
RV: right ventricle
RVOTO: right ventricular outflow tractobstruction
TR: tricuspid regurgitation
TV: tricuspid valve
Disclosure: Dr. Rafael Alonso-González, has received a research grantfrom Fundación Alfonso Martín Escudero, Madrid, Spain.
Professor Gatzoulis and the Royal Brompton Adult Congenital HeartDisease and Centre for Pulmonary Hypertension have received supportfrom the British Heart Foundation.
Correspondence: Prof M. A. Gatzoulis, MD, PhD, FACC,
Adult Congenital Heart Centre and Centre for Pulmonary Hypertension.Royal Brompton Hospital,
Sydney Street. SW3 6NP, London. United Kingdom
E-mail: m.gatzoulis@rbht.nhs.uk