Palabras clave
INTRODUCCIÓN
En comparación con el ventrículo izquierdo (VI),hasta hace relativamente poco tiempo se ha prestado poca atención al ventrículo derecho (VD) tanto en los pacientes con cardiopatías adquiridascomo, en menor medida, en los pacientes con cardiopatías congénitas (CC). En general, no está clarosi la disfunción del VD afecta al pronóstico de lospacientes con cardiopatías adquiridas; sin embargo,en los pacientes con CC parece que hay relaciónentre la disfunción y fibrosis del VD y la calidad devida, los síntomas y el pronóstico. En la mayoría delos casos, en los pacientes con CC el VD es el ventrículo subpulmonar; sin embargo, esto no siemprees así, como por ejemplo en los pacientes con transposición de grandes vasos, en los que el VD soportala circulación sistémica. Por lo tanto, la funciónventricular derecha puede verse deteriorada comoresultado de una sobrecarga de presión o de volumen o por la combinación de ambas. Así pues, elconocimiento exacto de la anatomía del VD y la determinación de sus volúmenes y su función son de capital importancia para los clínicos que atienden alos pacientes con CC. En este artículo se revisa laanatomía del VD y se examina el papel central quedesempeña en las CC.
ANATOMÍA
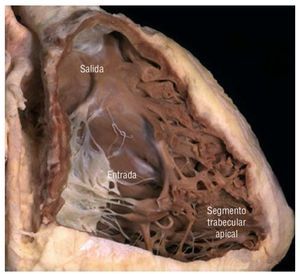
El VD está formado por tres segmentos diferentes: segmento de entrada, segmento apical trabecular y segmento de salida (fig. 1). El componentede entrada se extiende desde el anillo de la válvulatricúspide (VT) hasta la inserción de los músculospapilares en la pared ventricular; por lo tanto,rodea y soporta los velos y el aparato subvalvulartricuspídeos. La VT tiene tres velos: septal, antero-superior e inferior (o mural). Su característica másdistintiva es la presencia de cuerdas tendinosas queinsertan su velo septal en el tabique interventricular,lo cual, junto con su desplazamiento apical, permitediferenciarla de la válvula mitral y del ventrículomorfológicamente izquierdo en la ecocardiografía.Otro indicador útil para identificar el ventrículomorfológicamente derecho es la presencia de la banda moderadora, que es un grueso fascículo muscular que cruza la cavidad ventricular derecha.

Fig. 1. Esta disección muestra los tres componentes de un ventrículo derecho normal. Cortesía de la Prof. Y. Ho.
El componente apical trabecular incluye el cuerpoy el ápex del VD y se caracteriza por marcadas trabeculaciones. El segmento de salida es muscular yalargado, y se extiende hasta la válvula pulmonar,la cual carece de un verdadero anillo valvular. Estaforma más triangular del VD lo diferencia del VI,que es más cónico.
Ambos ventrículos se diferencian significativamente en la orientación de las fibras musculares.Mientras que en el VD las fibras musculares se disponen de forma circunferencial y longitudinal, lapared del VI se caracteriza por una orientaciónoblicua de sus fibras superficiales, mientras queestas se disponen longitudinalmente en el subendocardio y circunferencialmente en el miocardio,como si se tratase de un sandwich1. El espesornormal de la pared libre del VD es de 3-5 mm, peroen condiciones de sobrecarga de presión dicho espesor puede ser incluso superior al del VI, al igualque ocurre con su arquitectura muscular2.
EL VENTRÍCULO DERECHOCON SOBRECARGA DE PRESIÓN
Los dos modelos más frecuentes del VD con sobrecarga de presión son la obstrucción del tracto desalida del VD (TSVD) y el VD sistémico.
OBSTRUCCIÓN DEL TRACTO DE SALIDAVENTRICULAR DERECHO
La obstrucción congénita del TSVD se debe en lamayoría de los casos a una estenosis pulmonar(EP), pero también puede ser consecuencia de anomalías en la parte media del VD, el infundíbulo, laregión supravalvular o las ramas principales o periféricas de las arterias pulmonares3 (tabla 1).

Estenosis valvular pulmonar
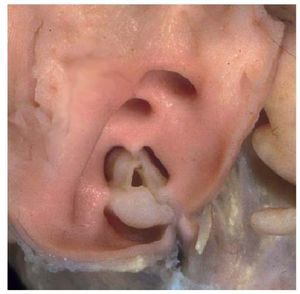
La EP aislada se da en un 80-90% del total de pacientes con obstrucción del TSVD, y casi siempre escongénita. Se puede identificar tres tipos morfológicos de válvula pulmonar estenótica: a) en formade cúpula; b) displásica, y c) bicúspide o unicúspide4 (poco frecuente). La forma clásica de la EP esla válvula pulmonar en forma de cúpula, la cual secaracteriza por tener entre 2 y 4 rafes, sin separación de las cúspides valvulares5 (fig. 2). Se caracteriza por ser una válvula móvil con una aperturacentral estrecha6. Aunque puede ser hereditaria, sutransmisión familiar es baja, y oscila entre el 1,7y el 3,6%7,8.

Fig. 2. Válvula pulmonar en forma de cúpula, vista desde el lado arterial.Cortesía de la Prof. Y. Ho.
La válvula pulmonar displásica está presente enun 10-20% de los pacientes con EP9,10. Las válvulas displásicas son válvulas tricúspides con velos engrosados, formados por tejido mixomatoso desorganizado y en general poco o nada fusionados. Su presencia suele asociarse a una unión ventrículo-arterialhipoplásica. Esta entidad es el hallazgo más frecuente en los pacientes con EP asociada al síndromede Noonan10.
La EP, cuando es significativa, da lugar a una hipertrofia compensatoria del VD, especialmente infundibular. Cuando la hipertrofia es prominente,puede producir una estenosis subvalvular dinámicasecundaria. La EP también puede producir una dilatación postestenótica del tronco de la arteria pulmonar, que es más frecuente en la EP con válvulapulmonar en forma de cúpula. Esta dilatación a menudo se extiende hasta la porción proximal de la arteria pulmonar izquierda, posiblemente debido a laorientación del chorro de alta velocidad a través deun orificio valvular estenótico. Anatómicamente, elchorro se dirige hacia la arteria pulmonar izquierda(continuación natural de la arteria pulmonar principal) y puede producir una distribución desigual delflujo sanguíneo, con mayor tendencia a dirigirse alpulmón izquierdo11. Además, la presencia de anomalías intrínsecas de la pared arterial pulmonar contribuye también a la dilatación de la arteria pulmonar.Es interesante señalar que la dilatación postestenótica de la arteria pulmonar es muy poco frecuente enpacientes con estenosis valvular displásica.
Un VD hipertrófico puede conservar su funcióndurante años, aun en presencia de presiones ventriculares derechas casi sistémicas. Es exageradoafirmar que el VD se dilata y falla a edades tempranas de la vida en presencia de presiones ventriculares derechas crónicamente elevadas. De hecho,en tanto se preserve el ritmo sinusal y no haya unasobrecarga de volumen adicional, el VD suele sercapaz de mantener su función sistólica hasta lacuarta o la quinta década de la vida12.
La mayoría de los pacientes con EP se mantienenasintomáticos durante años, incluso cuando la estenosis progresa de moderada a grave. Por consiguiente, no es infrecuente que el diagnóstico se hagaen la edad adulta. Tanto en la infancia como en laedad adulta, este trastorno suele detectarse al auscultar un soplo característico en la exploración física,y se confirma luego mediante ecocardiografía transtorácica. Los síntomas aparecen generalmentecuando la presión del VD supera el 50% de la presión sistémica, lo cual limita el gasto del VD y la perfusión pulmonar. La disnea de esfuerzo y la fatigason los síntomas más frecuentes. Puede aparecertambién dolor torácico, síncope o incluso muerte súbita cardiaca en pacientes con EP grave, y se cree queello se debe a una disminución de la perfusión miocárdica como consecuencia de un gasto cardiaco insuficiente durante el ejercicio, que produce isquemiamiocárdica y arritmias ventriculares3.
Los signos auscultatorios en la EP son bastantecaracterísticos; la válvula pulmonar móvil produceun clic de eyección que se reduce con la inspiración.Cuanto más grave es la estenosis, más temprana es la aparición del clic en sístole, hasta que llega a fusionarse con el primer ruido cardiaco y se hace inaudible. También hay un soplo sistólico eyectivo en crescendo-decrescendo, que es máximo en el bordesuperior esternal izquierdo. La insuficiencia pulmonar (IP) es infrecuente en este contexto. A menudo se ausculta un cuarto ruido cardiaco en elborde inferior esternal izquierdo en los pacientescon estenosis grave. Cuando hay un tercer ruidocardiaco, debe sospecharse una comunicación interauricular (CIA) asociada.
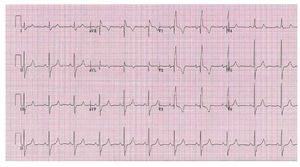
El electrocardiograma suele ser normal cuando lapresión sistólica del VD es < 60 mmHg. Cuandoaumenta la gravedad de la lesión, pueden aparecersignos electrocardiográficos de crecimiento auricular derecho, desviación del eje del QRS a la derecha e hipertrofia del VD13. En la EP grave, elpatrón habitual en las derivaciones precordiales derechas suele ser de R, RS o QR, con onda R> 20 mm (fig. 3) en dichas derivaciones.

Fig. 3. Paciente con estenosis pulmonargrave. El ECG muestra un bloqueo derama derecha e hipertrofia ventricular derecha.
La radiografía de tórax muestra a menudo unascaracterísticas distintivas, con un tamaño cardiaconormal y una arteria pulmonar central dilatada.Este signo puede estar ausente en los pacientes conválvulas displásicas. Un patrón vascular acentuadoen la base pulmonar izquierda (signo de Chen) enpacientes con válvula pulmonar en forma de cúpulase debe a un flujo sanguíneo preferencial. En la EPgrave, dicho patrón puede estar reducido11. La aurícula derecha y el VD pueden estar dilatados en presencia de descompensación ventricular derecha ocuando la EP se asocia a una CIA.
La ecocardiografía es el método diagnóstico deelección. El Doppler continuo es útil para obtenerlos valores de gradiente instantáneo máximo através del TSVD/válvula pulmonar. Sin embargo,diferenciar la estenosis valvular de la obstruccióndinámica subvalvular secundaria a una hipertrofiadel infundíbulo puede resultar muy difícil. La utilización del Doppler color y pulsado permite localizar el lugar de la obstrucción y, por lo tanto, diferenciar la EP valvular de la subvalvular (TSVD) yla supravalvular. La obstrucción dinámica subvalvular acompaña a menudo a la EP valvular grave yse caracteriza por un chorro de aceleración tardía,similar al de la obstrucción dinámica del tracto desalida del VI. La ecocardiografía también puedeaportar información sobre la movilidad valvular, eltamaño y la función del VD y la presencia de dilatación postestenótica. La resonancia magnética cardiaca (RMC) puede aportar información adicionalpara evaluar la EP y localizar la zona exacta de laobstrucción. Además, la RMC es el patrón de referencia en la cuantificación del tamaño y la funcióndel VD.
La primera operación de una EP se realizó en1948 y consistió en una comisurotomía de la válvula pulmonar. Las intervenciones posteriores —decomienzos de los años cincuenta— consistieron envalvulotomías cerradas mediante el empleo de dilatadores valvulares especialmente diseñados para laEP (intervención de Brock). Posteriormente, la valvulotomía abierta llevó a unos resultados significativamente mejores. Si la válvula pulmonar es displásica, puede ser necesaria una valvulotomía parcial ototal; si hay una hipoplasia anular o del tronco dela pulmonar, puede llegar a ser necesario un parchetransanular. Todas estas intervenciones conllevan,como consecuencia, diversos grados de IP, que elpaciente puede tolerar bien durante muchos años.Inicialmente, el VD se dilata, manteniendo la contractilidad y el volumen de eyección. Con el pasodel tiempo, el VD puede claudicar y los pacientespresentan síntomas como disnea, fatiga y arritmias.
El pronóstico de los pacientes con EP es excelente. En el Second Natural History Study of Congenital Heart Defects (Segundo Estudio de la Evolución Natural de las Cardiopatías Congénitas), no hubo una progresión significativa de la enfermedaden pacientes con un gradiente máximo < 20 mmHg.La mayoría de los pacientes con valores de gradiente máximo > 50 mmHg necesitaron una intervención14, mientras que la probabilidad de quefuera necesaria una reparación fue del 20% enquienes tenían un gradiente máximo de 25-49 mmHg.Estudios de seguimiento de pacientes tratados en lainfancia con una valvulotomía quirúrgica hanpuesto de manifiesto que la evolución de esta cohorte es excelente, con una tasa de mortalidad inferior al 5% después de 25 años de seguimiento14. Sinembargo, una tercera parte de los pacientes desarrolla una IP significativa que requiere reintervención en algún momento de su vida (tasa de reintervenciones de un 9-40%; media de tiempo hastala sustitución de la válvula pulmonar [SVP],33 años)15-17. La presencia de arritmias auriculares oventriculares, intolerancia al ejercicio o cardiomegalia en la radiografía de tórax en pacientes con unavalvulotomía pulmonar previa debe hacer sospecharuna IP. Las valoraciones subjetivas de la toleranciaal esfuerzo pueden no ser fiables en estos pacientesdebido al carácter crónico de la IP y la progresiónlenta de la enfermedad. Por ello, la prueba de esfuerzo con consumo de oxígeno es esencial paracuantificar objetivamente la intolerancia al esfuerzoy realizar un seguimiento adecuado de su evolución.Además, la realización de ecocardiografías seriadases importante para detectar disfunción ventricularderecha o una insuficiencia tricuspídea (IT) significativa, ante las cuales debe considerarse la SVP12.
Desde que en 1982 se introdujo la valvuloplastiapercutánea con balón18, esta técnica se ha convertido en el tratamiento de elección para los pacientescon una EP valvular clásica en forma de cúpula.Las guías del American College of Cardiology/American Heart Association recomiendan la valvuloplastia percutánea en pacientes asintomáticos conuna válvula pulmonar en forma de cúpula y un gradiente pulmonar máximo instantáneo > 60 mmHgo un gradiente medio > 40 mmHg. En pacientessintomáticos, la valvuloplastia percutánea está indicada cuando hay un gradiente instantáneo máximo> 50 mmHg o un gradiente medio > 30 mmHg19. Los resultados a largo plazo tras la valvuloplastiacon balón son excelentes20, con una tasa de reestenosis muy baja. Las reestenosis son más frecuentescuando inmediatamente después de la intervenciónhay un gradiente residual significativo21. El riesgode IP significativa después de una valvuloplastiapercutánea con balón es del 5%. En esta cohorte depacientes, una fracción de regurgitación pulmonar> 15% se ha asociado a un menor consumo de O2 pico en la prueba de esfuerzo con consumo de oxígeno22. La posibilidad de que la valvuloplastia percutánea se asocie a una tasa de reintervención porIP similar a la de la valvulotomía quirúrgica es porel momento tan sólo una especulación.
Ventrículo derecho con doble cámara
El VD con doble cámara (VDDC) con frecuenciase asocia a una comunicación interventricular(CIV). Se caracteriza por la presencia de bandasmusculares aberrantes hipertrofiadas que dividen lacavidad ventricular en una cámara proximal de altapresión y una cámara distal de baja presión (fig. 4). Esto contrasta con lo que ocurre en la obstruccióndel TSVD en la tetralogía de Fallot (TdF), en la quela obstrucción se produce en el infundíbulo.

Fig. 4. Paciente con un ventrículo derechode doble cámara. La flecha muestra labanda muscular hipertrofiada.
La gravedad de la obstrucción en el VDDC tiende a progresar con el paso del tiempo, aunque es posible que no se manifieste hasta la edad adulta. Desde el punto de vista clínico, los pacientes con VDDC y tabique interventricular intacto se parecen a los pacientes con una EP aislada. En caso de CIV,las manifestaciones clínicas en la fase inicial de laenfermedad pueden estar dominadas por la CIV, envez de por el VDDC.
En la exploración física, es característico un soplopansistólico intenso, de tipo crescendo-decrescendo, que a menudo se acompaña de frémito y puede serindistinguible del producido por una EP aisladaaunque, a diferencia de esta, no hay clic de eyección. En presencia de una CIV, es posible auscultarun soplo característico relacionado con ella, amenos que la CIV desemboque en la cámara de altapresión (proximal).
El electrocardiograma habitualmente muestra hipertrofia del VD, lo cual no es común en los casosde CIV restrictiva aislada. La ecocardiografía bidimensional suele ser diagnóstica tanto para identificar el grado y la localización de la obstruccióncomo para descartar una CIV asociada, aunque esimportante tener en cuenta que la CIV puede ser difícil de visualizar cuando desemboca en la cámarade alta presión. La RMC aporta información complementaria respecto a la anatomía y la fisiología.
Las indicaciones para el tratamiento quirúrgicodel VDDC son similares a las de la EP valvular19. Las intervenciones de resección muscular y agrandamiento del tracto de salida han resultado muyefectivas, con excelentes resultados a largo plazo ytasas de recidiva muy bajas23.
Estenosis pulmonar y embarazo
Cuando la función del VD está preservada, laobstrucción aislada del TSVD suele ser bien tolerada durante el embarazo, a pesar de que la estenosis sea grave. No obstante, en caso de arritmiasauriculares recurrentes y/o una insuficiencia cardiaca derecha temprana durante el embarazo en pacientes con una EP grave, debe considerarse la posibilidad de realizar una valvuloplastia percutánea.
VENTRÍCULO DERECHO SISTÉMICO
El ventrículo morfológicamente derecho en posición sistémica en la edad adulta se encuentra, lamayor parte de las veces, en pacientes con unatransposición de grandes vasos corregida congénitamente (TGVcc) o en pacientes con una transposición de grandes vasos (TGV) tras una corrección fisiológica mediante switch auricular (operación deMustard o Senning). En posición sistémica, el VDmodifica la arquitectura de sus fibras musculares ypasa a asemejarse al patrón en sandwich que se observa en el VI normal4. Además, en el VD sistémicoel acortamiento de la pared libre es predominantemente circunferencial, en vez de longitudinal5. Estoscambios permiten al VD adaptarse en gran mediday durante varias décadas a la posición y la funciónsistémicas.
Transposición corregida congénitamente
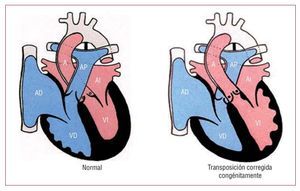
La TGVcc es un trastorno muy poco frecuente,que supone menos del 1% del total de CC. Consisteen una discordancia auriculoventricular (AV) yventriculoarterial (VA): la aurícula derecha está conectada al ventrículo morfológicamente izquierdo,el cual da origen a la arteria pulmonar, mientrasque la aurícula izquierda está conectada al ventrículo morfológicamente derecho, el cual da origen ala aorta. La aorta se encuentra generalmente,aunque no siempre, anterior y a la izquierda de laarteria pulmonar, y los grandes vasos suelen ser paralelos, en lugar de cruzarse (fig. 5). Es frecuente laasociación de esta entidad con anomalías en el situs. De hecho, la TGVcc es una de las CC que se asociacon más frecuencia a dextrocardia y debe sospecharse cuando esta se asocia con situs solitus abdominal. En un 95% de los casos de TGVcc hay también lesiones cardiacas asociadas, entre las que seencuentran la anomalía de Ebstein de la VT (90%),la CIV (70%), la EP (40%) y el bloqueo AV completo (un 2% de riesgo al año)24.

Fig. 5. Corazón normal y transposición corregida congénitamente. A: aorta; AD: aurícula derecha; AI: aurícula izquierda; AP: arteria pulmonar; VD: ventrículo derecho; VI: ventrículo izquierdo. Cortesía de la Prof. Y. Ho.
Es característico que los pacientes con TGVcc semantengan asintomáticos hasta la tercera o lacuarta década de la vida, si bien pueden tener síntomas en edades más tempranas cuando se asociacon otras lesiones hemodinámicamente significativas25. Los síntomas más frecuentes son la disneade esfuerzo y las palpitaciones o el síncope secundarios a arritmias auriculares o a un bloqueo AVcompleto. Los pacientes con CIV y EP asociadaspueden presentar cianosis progresiva.
La ecocardiografía es el método diagnóstico deelección. Las proyecciones apical de cuatro cámarasy subcostal son las más útiles para determinar tantoel situs cordis como las conexiones AV-VA. Lamejor forma de evaluar la morfología ventricular esmediante el examen de las válvulas AV (la VT estásiempre ligeramente desplazada hacia el ápex en relación con la válvula mitral, y su velo septal tieneinserciones de las cuerdas tendinosas en el tabiqueinterventricular), así como identificar la banda moderadora y la presencia de trabéculas que determinan cuál es el VD. La relación de los dos ventrículos y de los grandes vasos es más laterolateral que la posición anteroposterior habitual. Es importanteidentificar posibles lesiones asociadas como unaCIV, que suele ser perimembranosa, así como la presencia de una obstrucción del tracto de salida del VI,que puede deberse a un aneurisma del septo membranoso, una membrana fibrosa o «tissue tags» móviles a nivel subpulmonar. Además, en los pacientescon TGVcc es de crucial importancia realizar unaevaluación adecuada de la función del VD sistémico,así como del grado de insuficiencia de la válvula AVsistémica. La RMC proporciona una informacióncomplementaria sobre la anatomía y una estimaciónmás exacta del tamaño y la función del VD.
El pronóstico de la TGVcc depende en gran medida de la presencia de lesiones asociadas, así comode la gravedad de estas. Si no las hay, los pacientescon TGVcc pueden sobrevivir hasta la séptima o laoctava década de la vida26. Sin embargo, la incidencia de disfunción sistólica del ventrículo sistémico e insuficiencia cardiaca congestiva aumentacon la edad, incluso en ausencia de lesiones asociadas (más de una tercera parte de los pacientes sufrirán insuficiencia cardiaca congestiva en la quintadécada de la vida)27. La presencia de una IT significativa y/o una disfunción del VD sistémicose asocia con una mortalidad significativamentesuperior27,28 y con un mayor riesgo de sufrir insuficiencia cardiaca descompensada. La rapidez del deterioro de la función del VD sistémico en presenciade una IT significativa es muy superior a la que presenta el VI en posición sistémica en presencia de insuficiencia mitral. Sin embargo, continúan sin estarclaros los factores que explican este hecho. Es probable que la geometría ventricular desempeñe unpapel importante, puesto que una dilatación significativa del VD conlleva una dilatación del anillo tricuspídeo, lo que agrava más rápidamente la IT y causa a su vez disfunción ventricular derecha29. Porotro lado, también es probable que la perfusiónmiocárdica sea insuficiente para suplir la demandade un VD sistémico con una hipertrofia importante,lo que produciría un desajuste entre perfusión y demanda y, como resultado, isquemia miocárdica30. El hecho de que la función ventricular derecha sedeteriore en presencia de insuficiencia significativade la válvula AV sistémica, en especial si dicha válvula tiene anomalías intrínsecas, hace que sea recomendable considerar la sustitución de la VT antesde que se produzca una disfunción irreversible delVD sistémico19.
Aunque hay evidencias que indican una activación neurohormonal en los pacientes con TGVcc, espoco lo que se sabe acerca de la eficacia del tratamiento clásico para la insuficiencia cardiaca, comolos inhibidores de la enzima de conversión de angiotensina (IECA) o los bloqueadores beta (BB), enesta enfermedad31. Los datos existentes al respectoson contradictorios y muchos estudios han analizado conjuntamente a pacientes con TGVcc e intervenciones de Mustard o Senning; estos pueden responder de manera diferente a los IECA a causa deque tienen una «función» AV anormal. Un pequeñoestudio piloto realizado en pacientes con TGVcc oSenning indica que el carvedilol puede mejorar lafunción del VD sistémico. Sin embargo, los BBdeben usarse con precaución debido a la propensión a anomalías del sistema de conducción y el bloqueo AV completo32, a menos que el paciente tengaun marcapasos.
Transposición completa de grandes vasos
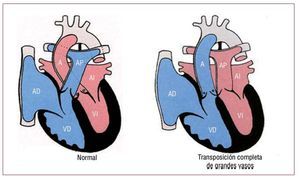
La transposición completa de grandes vasos se daen aproximadamente un 5% del total de recién nacidos con malformaciones congénitas cardiacas. Enestos casos, la aorta tiene su origen en el ventrículomorfológicamente derecho y la arteria pulmonar,en el ventrículo morfológicamente izquierdo (fig. 6).Los grandes vasos tienen un trayecto paralelo, conla aorta en posición anterior y generalmente a la derecha de la arteria pulmonar. Un tercio de los pacientes presentan lesiones asociadas.

Fig. 6. Corazón normal y transposición de grandes vasos. A: aorta; AD: aurícula derecha; AI: aurícula izquierda; AP: arteria pulmonar; VD: ventrículo derecho; VI: ventrículo izquierdo. Cortesía de la Prof. Y. Ho.
En la actualidad, el tratamiento de elección paralos pacientes con TGV es la corrección anatómica o switch arterial, que viene realizándose desde hacemás de dos décadas. Sin embargo, a la mayor partede los adultos vivos con TGV se les ha practicadouna corrección fisiológica o switch auricular (operación de Mustard o de Senning), tras la cual el VDsoporta la circulación sistémica. Estos pacientespueden sufrir una disfunción ventricular sistémica,arritmias o problemas relacionados con los drenajesauriculares, y ello justifica un seguimiento anual encentros especializados en cardiopatías congénitasdel adulto (CCA)19. En estos pacientes la funciónventricular derecha puede sufrir un deterioro progresivo, lo cual puede asociarse con un agravamiento de la IT. Los pacientes con una IT significativa debida a una enfermedad primaria de la VT(que puede ser iatrogénica tras el cierre de una CIVo debida a otros factores como una endocarditis)pueden beneficiarse de una sustitución valvular19. La evaluación de la función del VD sistémico,aunque difícil, es de capital importancia. En estecontexto, la evaluación ecocardiográfica se limita ala obtención de datos semicuantitativos, pero enmanos expertas puede proporcionar informaciónhemodinámica clave sobre el VD sistémico. Se hademostrado que el desplazamiento del eje largo dela pared libre del VD sistémico predice la capacidadde ejercicio en esos pacientes33. La ecocardiografíatambién proporciona información sobre la permeabilidad de los drenajes sistémico y pulmonar,así como de la presencia de CIA residuales o insuficiencia valvular.
La RMC es la técnica de referencia para el estudio del tamaño y la función del VD en esta población y permite evaluar con mayor exactitud los drenajes venosos pulmonar y sistémico. Cuando laRMC está contraindicada, la angio-TC con sincronización electrocardiográfica puede ser una alternativa.
Tanto las bradiarritmias como las taquiarritmiastardías son frecuentes tras una intervención de switch auricular. La disfunción del nodo sinusal escomún, y un 11% de los pacientes necesitan un marcapasos en algún momento de la vida34. Al menosen una cuarta parte de los pacientes se desarrollaflutter o fibrilación auriculares en un plazo de 23años tras la intervención de switch auricular35. Laamplia cicatrización auricular que se produce trasel switch auricular constituye el sustrato perfectopara la aparición de arritmias auriculares, quepueden poner en peligro la vida del paciente cuandoson rápidas o si hay una obstrucción significativade alguno de los drenajes auriculares. Los fármacosantiarrítmicos con efecto inotrópico negativo se utilizan con muy poca frecuencia en presencia de disfunción del VD sistémico. La ablación con radiofrecuencia de las arritmias auriculares puede ser unverdadero reto técnico, pero su tasa de éxito es deaproximadamente un 70% en manos expertas36.
La obstrucción de los drenajes sistémico o pulmonar o la presencia de comunicaciones auricularesresiduales, aunque son infrecuentes, deben descartarse siempre en pacientes sintomáticos. La obstrucción de la vena cava superior (VCS) es la máscomún, si bien es infrecuente que se produzca un«síndrome de VCS», puesto que generalmente el sistema de la vena ácigos se dilata y compensa la obstrucción. La permeabilidad del drenaje venoso sistémico superior debe evaluarse siempre antes deimplantar un marcapasos intracavitario. Las comunicaciones auriculares residuales son relativamentemás frecuentes. Generalmente son pequeñas y carecen de trascendencia hemodinámica, pero puedenser el sustrato para que se produzcan embolias paradójicas y pueden justificar una cianosis en reposoo durante el ejercicio. Cuando hay una dilatacióndel VI, debe realizarse una ecocardiografía con contraste, mediante el empleo de suero fisiológico agitado, con objeto de descartar una comunicaciónauricular significativa, con un cortocircuito predominantemente sistémico-pulmonar (equivalente aun crecimiento del VD en un corazón concordantecon una comunicación auricular).
Aproximadamente un 7% de los pacientes sufrenhipertensión arterial pulmonar tras el switch auricular para la TGV37,38. La patogenia se desconoce, aunque existen algunos factores de riesgo conocidos, como que la intervención quirúrgica se realice después de los 2 años de edad39 y la presencia deun cortocircuito ventricular o en los grandes vasosantes de la reparación40. En presencia de hipertensión pulmonar, debe descartarse la posible obstrucción del drenaje venoso pulmonar como su causa.
Ventrículo derecho sistémico y muerte súbitacardiaca
La mayoría de los pacientes con un VD sistémicofallecen de forma súbita41,42. La edad, la disfunciónventricular sistémica, la clase funcional de la NewYork Heart Association43, las arritmias supraventricularves35 y la dispersión del QT44 son factores que sehan asociado con muerte súbita en estos pacientes.Recientemente, Schwerzmann et al43 han descritoque una duración del QRS ≥ 140 ms se asocia a unapeor clase funcional, una peor función del VD sistémico y una mortalidad más elevada. Recientementese han utilizado desfibriladores automáticos implantables (DAI) en este contexto, aunque su papel todavía no está claramente establecido.
Ventrículo derecho sistémico y embarazo
El riesgo que comporta el embarazo en las pacientes con un VD sistémico depende de la funciónventricular sistémica, la presencia de lesiones hemodinámicas significativas y la capacidad funcional45. El embarazo puede producir un deterioro de la función del VD sistémico, pero continúan sin estarclaras sus repercusiones a largo plazo en la funciónventricular derecha. Se ha descrito una tasa de mortalidad de hasta un 4% en una pequeña serie de mujeres embarazadas con VD sistémico, y este hechodebe comentarse con las pacientes en el consejo previo a la concepción46. En las pacientes con unafunción sistólica del VD buena o ligeramente deprimida que no tengan una obstrucción significativade ninguno de los drenajes auriculares, el riesgo delembarazo es relativamente bajo47,48. Durante el embarazo, la evaluación clínica debe centrarse en lossignos iniciales de insuficiencia cardiaca y de arritmias. Si se produce una taquicardia auricular,puede realizarse de forma segura una cardioversióneléctrica para restablecer el ritmo sinusal. Debeconsiderarse el uso de ácido acetilsalicílico durantetodo el embarazo en las pacientes con antecedentesde arritmias auriculares.
EL VENTRÍCULO DERECHO CON SOBRECARGA DE VOLUMEN
Las tres lesiones más frecuentes asociadas a la sobrecarga de volumen del VD son la CIA, la IP en elcontexto de la TdF y la IT en el contexto de la anomalía de Ebstein. En esta revisión nos centraremosen los dos últimos.
TETRALOGÍA DE FALLOT
La TdF es la forma más frecuente de CC cianóticadespués del primer año de vida. Consiste en una CIVsubaórtica grande con una mala alineación del septointerventricular, un acabalgamiento de la aorta sobreel defecto del tabique interventricular (que cuando esmayor del 50% se clasifica como VD de doble salida), obstrucción del TSVD e hipertrofia del VD.Puede haber una estenosis o una hipoplasia de ramaspulmonares, y algunos pacientes presentan unaatresia pulmonar. Las anomalías asociadas más frecuentes son una CIA, un canal AV (más frecuente enpacientes con síndrome de Down) y un arco aórticoderecho, que puede darse en hasta un 25% de los pacientes. Puede haber también anomalías de las arterias coronarias, que afectan con frecuencia a la arteria coronaria descendente anterior izquierda, quetiene su origen en el seno coronario derecho y cruzael TSVD (un 3% de los casos)49.
La mayor parte de los pacientes con TdF se someten a una reparación total en la infancia. Elloconlleva el cierre de la CIV y la resolución de laobstrucción del TSVD. Esto último puede requerirla utilización de un parche transanular/TSVD, quealtera la integridad del «anillo» de la válvula pulmonar, o una valvulotomía/valvectomía si la válvula pulmonar es anormal. Puede ser necesario utilizar un conducto extracardiaco entre el TSVD y laarteria pulmonar principal en casos con atresia pulmonar o un trayecto anómalo de la arteria coronaria izquierda que cruce el TSVD.
La secuela más frecuente tras la reparación de laTdF es la aparición de una IP significativa. La IP grave crónica puede causar dilatación y disfunciónsistólica del VD50, con propensión a la aparición dearritmias clínicas y muerte súbita cardiaca51,52. Se sabe que después de dos décadas de exposición auna sobrecarga de volumen significativa la funciónsistólica del VD sufre un deterioro que da lugar auna intolerancia progresiva al esfuerzo y un riesgoelevado de arritmias, tanto supraventriculares comoventriculares. Por consiguiente, en estos pacientesdebe considerarse el recambio valvular pulmonaren caso de dilatación significativa del VD, antes deque aparezca una disfunción ventricular irreversible. Basarse únicamente en los síntomas para decidir el momento adecuado para practicar la SVPpuede llevar a error, puesto que es frecuente que lossíntomas aparezcan cuando la función del VD yaesté gravemente deteriorada. La evaluación objetivade la capacidad de ejercicio puede ser más fiable, ylas pruebas de esfuerzo con consumo de oxígeno seriadas pueden ser útiles para identificar cambios enla capacidad de ejercicio que el paciente puede nohaber percibido. La intervención quirúrgica está indicada también cuando los pacientes con una IPgrave y dilatación del VD desarrollan una IT moderada o grave, o arritmias auriculares o ventricularessintomáticas19. De hecho, no hay que tener encuenta un único parámetro, sino varias variables,para decidir el momento adecuado para la SVP.
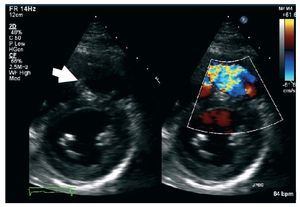
La ecocardiografía continúa siendo la modalidadde diagnóstico por imagen más ampliamente utilizada para evaluar a los pacientes con TdF. Sin embargo, la RMC es el patrón de referencia como técnica para la cuantificación de la IP y el análisisvolumétrico del VD. En los pacientes con una TdFreparada, y especialmente en los que presentan unaIT progresiva u otras secuelas, debe efectuarse unaecocardiografía anual y una RMC cada 2-3 años19. La cuantificación de la IP mediante la ecocardiografía puede resultar difícil para un operador pocoexperimentado. La igualación temprana de las presiones del VD y la arteria pulmonar puede ser unsigno indicativo de una IP significativa en ausenciade un VD restrictivo. Un tiempo de hemipresión< 100 ms53 y un índice de regurgitación pulmonar(cociente entre la duración del IP y el tiempo diastólico total) < 0,77 tiene una sensibilidad del 100%y una especificidad del 85% para identificar a pacientes con una fracción de regurgitación pulmonarsuperior al 25% en la RMC54 (fig. 7). Es necesarioevaluar también el tamaño y la función del VD.

Fig. 7. Paciente con tetralogía de Fallot reparada e insuficiencia pulmonar grave. El índice de regurgitación pulmonar es del 58% (b/a).
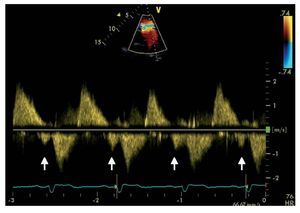
La identificación mediante Doppler pulsado deun flujo pulmonar diastólico tardío anterógrado,que coincide con la sístole auricular, está presentedurante todo el ciclo respiratorio y se asocia a unflujo retrógrado en la vena cava inferior, que definela denominada «fisiología restrictiva del VD» eneste contexto (fig. 8). Dicha fisiología con frecuencia se da tempranamente tras la reparación dela TdF, y se ha demostrado que se asocia a un bajogasto cardiaco y una estancia en la unidad de cuidados intensivos más larga55,56. Sin embargo, hayindicios de que la fisiología restrictiva del VD enuna fase tardía tras la reparación de la TdF seasocia a menor tamaño del VD, menor duración delQRS y mejor capacidad de ejercicio, posiblementepor contrarrestar los efectos de la IP, lo que contribuye a mejorar el flujo pulmonar anterógrado y,por lo tanto, el gasto cardiaco57,58.

Fig. 8. Paciente con tetralogía de Fallot reparada y ventrículo derecho restrictivo. Las flechas indican el flujo diastólico anterógrado tardío (onda «a»).
La forma poco habitual que tiene la cavidad delVD y el modo impredecible en que se dilata hacenque el análisis cuantitativo exacto mediante técnicasecocardiográficas o de angiografía radioisotópicaresulte difícil. La RMC permite un análisis cuantitativo más exacto y reproducible de las dimensionesy la función del VD en manos experimentadas. El«valor de corte» óptimo a partir del cual debe recomendarse la SVP continúa siendo objeto de controversia. Therrien et al59 indicaron que los volúmenesdel VD no se normalizan en los pacientes con unvolumen telediastólico preoperatorio del VD> 170 ml/m2 y un volumen telesistólico del VD> 85 ml/m2. Oosterhof et al60 han señalado que nohay un valor umbral por encima del cual los volúmenes del VD no disminuyan tras la intervenciónquirúrgica, pero la normalización de los volúmenesdel VD sólo puede alcanzarse cuando el volumentelediastólico del VD no supera el valor de 160 ml/m2 o el volumen telesistólico del VD no es > 82 ml/m2. Recientemente se ha demostrado que una SVPtemprana en pacientes con un volumen telediastólico indexado del VD de 150 ml/m2 da lugar a unanormalización de las dimensiones del VD61, y muchos autores lo consideran un valor de corte,aunque es preciso tener en cuenta otros parámetros.
La mayor parte de los pacientes a los que se hapracticado una reparación de la TdF presentan enel electrocardiograma un bloqueo de rama derechajunto con signos de hipertrofia del VD. El aumentode duración del QRS tiene una buena correlacióncon la dilatación y la disfunción progresiva del VD.Se cree que hay una interacción eléctrico-mecánica,de forma que una mayor dilatación ventricular implica un retraso de la conducción y un aumento dela probabilidad de arritmias ventriculares malignas52. En consecuencia, el seguimiento de la prolongación del QRS a lo largo del tiempo es importante, de tal manera que un aumento en la duracióndel QRS ≥ 4 ms/año se asocia a un aumento delriesgo de taquicardia ventricular sostenida y muertesúbita cardiaca51. Otros factores predictivos de taquicardia ventricular y muerte súbita son una duración del QRS ≥ 180 ms y la disfunción del VI51,52, a pesar de que esta tiene un valor predictivo positivobajo cuando se considera aisladamente (el 29 frenteal 93% cuando se considera conjuntamente con laduración del QRS)62. La estratificación del riesgo demuerte súbita sigue siendo un reto en pacientes conTdF, y el papel de la implantación de un DAI paraprevención primaria en pacientes en alto riesgo(aneurismas grandes del TSVD63, QRS > 180 ms52,taquicardia ventricular inducible64, captación tardíade gadolinio65 significativa o disfunción del VI62)sigue sin estar claro66. Se ha demostrado que lasarritmias auriculares y la reducción de la variabilidad de la frecuencia cardiaca son también un indicador del aumento de morbilidad y mortalidad enesta población.
Tetralogía de Fallot y embarazo
Aunque las pacientes con insuficiencias valvulares toleran bien el embarazo, se ha descrito que laIP grave tras la reparación de una TdF es un factorde riesgo para la gestante67. Además, el riesgo materno de episodios clínicos sintomáticos asociadosal embarazo aumenta en presencia de disfunción delVD o el VI o de una hipoplasia de las arterias pulmonares. Sin embargo, esto último es infrecuente.El embarazo comporta un riesgo relativamente bajopara la mayoría de las mujeres con una TdF reparada, y durante la gestación las evaluaciones periódicas deben centrarse en los signos de disfuncióncardiaca derecha e izquierda y en la presencia dearritmias clínicas68.
ANOMALÍA DE EBSTEIN
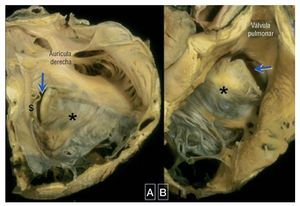
La anomalía de Ebstein es una CC muy infrecuente y compleja que supone menos del 1% del totalde casos de CC. Se caracteriza por la adherencia delos velos septal y posterior de la VT al miocardiosubyacente a causa de la ausencia de su deslaminación durante el desarrollo embrionario. Ello da lugara un desplazamiento apical del anillo funcional de laVT (velo septal más que el posterior y más que el anterior). El velo anterior puede estar gravemente deformado y formar una cortina intracavitaria extensaen forma de vela que puede llegar a producir unaobstrucción del TSVD (fig. 9). El segmento de entrada del VD está integrado funcionalmente a la aurícula derecha («VD atrializado») y puede llegar apresentar una dilatación desproporcionada, con diversos grados de adelgazamiento de la pared69. Estasanomalías anatómicas y funcionales pueden causaruna IT significativa, que conduce a una dilatación dela aurícula derecha y del VD. Es característico que elVI sea pequeño («comprimido») y con frecuenciatiene una función intrínsecamente anormal.

Fig. 9. Malformación de Ebstein de la válvula tricúspide vista desde la aurícula derecha (A) y desde el tracto de salida ventricular derecho (B). En este corazón, el velo anterior en forma de vela (*) tiene un margen libre limitado. Esto produce una apertura de llave-cerradura (flecha) entre la valva anterior y la valva septal subdesarrollada (s) que constituye el orificio valvular efectivo desplazado hacia el tracto de salida. Cortesía de la Prof. Y. Ho.
La anomalía de Ebstein puede asociarse a diferentes cardiopatías congénitas, como CIA, que seda en un 80-94% de los pacientes, EP, atresia de laválvula pulmonar o hipoplasia de arterias pulmonares, estenosis subaórtica, válvula aórtica bicúspide, prolapso de la válvula mitral y CIV. Además,los pacientes con anomalía de Ebstein tienen mayorprevalencia de vías accesorias (síndrome de Wolff-Parkinson-White).
La primera clasificación de la anomalía de Ebstein fue la propuesta por Carpentier et al70 en 1988.En ella se establecían cuatro categorías:
- Tipo A: el volumen del VD verdadero es adecuado.
- Tipo B: hay un gran componente atrializado delVD, pero el velo anterior se desplaza libremente.
- Tipo C: el movimiento del velo anterior estámuy limitado, lo que puede causar una obstrucciónsignificativa del TSVD.
- Tipo D: atrialización casi completa del VD, excepto por un pequeño componente infundibular.
Esta clasificación, aun siendo bastante simple,continúa siendo útil para decidir el tipo de cirugía arealizar.
La Glasgow Outcome Scale ampliada71 es unaclasificación más reciente y consiste en un sistemade puntuación mediante ecocardiografía que aportainformación pronóstica. Se compara el cociente delárea conjunta de la aurícula derecha y el «VD atrializado» con la del VD funcional y la del corazón izquierdo (grado 1, < 0,5; grado 2, 0,5-0,99; grado 3,1-1,49; grado 4, ≥ 1,5). Los pacientes que se encuentran en el grado 4 tienen más riesgo de mortalidad,mientras que la probabilidad de supervivencia másallá de la edad infantil es del 92% en aquellos conun cociente < 1. Un grado entre 1 y 4 se relacionacon una menor mortalidad, si bien en este grupo lamortalidad en la fase inicial de la infancia puede ascender hasta el 45%.
La forma de presentación clínica de la anomalíade Ebstein depende de la gravedad de la IT, la función del VD, el tamaño de la aurícula derecha, lapresión de la aurícula derecha y la presencia de uncortocircuito derecha-izquierda. Aunque la mayoríade los pacientes con anomalía de Ebstein sobreviven a la infancia, pueden presentar insuficiencia cardiaca grave y cianosis en edades tempranas de lavida. Sin embargo, estos síntomas suelen mejorar amedida que las resistencias vasculares pulmonaresdisminuyen. La intolerancia al ejercicio, con disneay/o fatiga, arritmias sintomáticas e insuficienciacardiaca derecha (congestiva) son los signos o síntomas de presentación más frecuente en la edadadulta. Generalmente, cuanto menor es la edad enel momento de la presentación clínica, más grave esla alteración anatómica y hemodinámica. Cuandohay una CIA o un foramen oval permeable, puedeaparecer cianosis en reposo o durante el ejercicio ypueden producirse embolias paradójicas. Aunquepoco frecuente, puede haber un cortocircuito interauricular de izquierda a derecha en pacientes conuna IT leve y aumento ligero de la presión de la aurícula derecha, lo cual puede contribuir a su vez a ladilatación del VD.
Las taquiarritmias son un modo de presentaciónde la anomalía de Ebstein más frecuente que la insuficiencia cardiaca en los adultos y afectan a un20-30% de los pacientes72. Están relacionadas con elcrecimiento de las cámaras cardiacas derechas ocon la presencia de una o varias vías accesorias deconducción.
En la exploración física, la presión venosa yugular rara vez está elevada en la anomalía de Ebstein, ni siquiera en presencia de una IT grave, dadoel notable agrandamiento de la aurícula derecha,que no permite la propagación del flujo de regurgitación hacia la vena cava superior. Puede haber cianosis periférica, sobre todo cuando existe un cortocircuito interauricular de derecha a izquierda y unaanomalía de Ebstein grave y/o bajo gasto cardiaco.Con frecuencia el VD es palpable en la región precordial y el volumen de pulso puede ser bajo. A laauscultación, el primer ruido cardiaco es intenso, ypuede haber un clic sistólico relacionado con unvelo anterior de la VT agrandado, en forma de vela.Puede auscultarse un soplo holosistólico en presencia de una IT significativa; este soplo puede aumentar con la inspiración, pero solamente cuandohay un VD adecuado funcional.
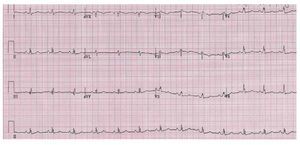
El electrocardiograma es anormal en la mayorparte de los pacientes con anomalía de Ebstein. Seobserva una onda P alta y ancha como resultadodel agrandamiento auricular derecho. Es característica la presencia de un bloqueo la rama derecha,completo o parcial. El QRS en las derivaciones precordiales del lado derecho del tórax puede ser debajo voltaje (fig. 10). Puede haber también vías accesorias y un bloqueo AV de primer grado.

Fig. 10. Paciente con anomalía de Ebstein. El ECG muestra un QRS de bajo voltaje y ondas P altas.
La silueta cardiaca puede variar entre el aspectonormal y un agrandamiento importante, con un corazón en forma de globo. La vascularidad pulmonar puede ser normal o estar disminuida. Un índice cardiotorácico > 0,65 se asocia a malpronóstico.
La ecocardiografía permite realizar una evaluación exacta de los velos de la VT, así como del tamaño y la función de las cámaras cardiacas derechas e izquierdas. El signo ecocardiográficoprincipal de la anomalía de Ebstein es el desplazamiento apical de los velos septal y posterior de laVT, que supera los 20 mm o los 8 mm/m2 de superficie corporal en los adultos73. También puede valorarse la localización y el grado de IT, así como lafactibilidad de una reparación de la VT. Hay quetener cuidado de no subestimar la gravedad de la ITcuando es grave («libre»), puesto que el flujo transtricuspídeo puede ser laminar, con una velocidadbaja y un perfil Doppler con un pico máximo temprano12. La RMC puede aportar información adicional importante en cuanto a la estructura y lafunción del corazón, que es esencial para una adecuada valoración preoperatoria.
La elección del momento apropiado para la reparación/sustitución de la VT es importante, y la gravedad de la anomalía de Ebstein, la disfunción delVD y el VI y la clase funcional son factores predictivos de la evolución posterior. Anteriormente, uncociente cardiotorácico > 65% se consideraba una indicación quirúrgica. Sin embargo, actualmente seacepta de manera general que debe ofrecerse el tratamiento quirúrgico antes de que se produzca unacardiomegalia importante, puesto que ello se asociaa una dilatación grave del VD y una disfunción sistólica grave. Muchos autores consideran una buenaindicación para la cirugía la combinación de los siguientes factores: síntomas debilitantes crecientes odeterioro gradual de la capacidad de ejercicio (cono sin cianosis), embolia paradójica, cardiomegaliaprogresiva en la radiografía de tórax y dilataciónprogresiva del VD o deterioro de la función sistólica del VD19. Aunque no se ha determinado si seobtiene un mejor resultado a largo plazo con la reparación o la sustitución de la VT, siempre que seaposible es preferible la reparación. La realizaciónsimultánea de una intervención quirúrgica para laarritmia en los pacientes con antecedentes de arritmias auriculares puede aportar un efecto beneficioso adicional.
Anomalía de Ebstein y embarazo
Pueden tolerar bien el embarazo las mujeres nocianóticas con anomalía de Ebstein, con o sin reparación previa, si están asintomáticas o mínimamente sintomáticas. Sin embargo, se ha descrito unaumento del riesgo de complicaciones del embarazoy pérdida fetal en estas mujeres, sobre todo en presencia de cianosis y/o una IT grave sintomática oarritmias74. Ante tales signos/lesiones, debe considerarse la reparación previa al embarazo. La tasa derecurrencia de la CC descrita en los hijos de pacientes con anomalía de Ebstein es de aproximadamente un 4%75.
TERAPIA DE RESINCRONIZACIÓN CARDIACAY VENTRÍCULO DERECHO
La terapia de resincronización cardiaca (TRC)mejora los parámetros hemodinámicos y la capacidad funcional y reduce la morbimortalidad en lospacientes con una insuficiencia cardiaca adquirida76. Sin embargo, hay poca evidencia que respalde el uso de la TRC en pacientes con CCA. Hayalguna evidencia de que la TRC puede mejorar deforma aguda la hemodinámica y facilitar la posibilidad de desconexión del bypass cardiorrespiratorioen este contexto. Janousek et al77 demostraron quela TRC mejora la función del VD sistémico en lospacientes con un retraso electromecánico nativo oinducido por un marcapasos intracavitario en elventrículo subpulmonar (VI). Sin embargo, la implantación de un resincronizador en pacientes conTGVcc o TGV puede resultar difícil, dada la anatomía del seno coronario y de las venas coronarias78. Además, aun cuando resulte factible, es difícil decidir en qué pacientes se obtendrá un efecto beneficioso con este tratamiento y parece claro que senecesitan más estudios en este campo.
La asincronía biventricular puede producirse también en pacientes con un bloqueo de rama derechatras la reparación de una TdF, y con frecuencia seasocia a una reducción de la función general y regional del VI79. Sin embargo, no está claro de quéforma puede aplicarse con éxito la TRC en este contexto. Por otra parte, la presencia de áreas de activación tardía del VD en la pared libre80, en zonas deltabique interventricular80 y en el tracto de salida81 implica probablemente que la diana exacta para laresincronización del VD difiere de un paciente aotro. En la actualidad la evidencia disponible no essuficiente para recomendar el uso sistemático de laTRC en esta población y son necesarios más datos.
CONCLUSIONES
El VD, con su compleja geometría y sus mecanismos de adaptación únicos en las CC, continúaplanteando un verdadero reto a los cardiólogos deadultos. El mantenimiento de una función adecuada del VD y evitar la dilatación excesiva de esteson esenciales tanto si se encuentra en una posiciónsubpulmonar como si tiene una posición sistémica,e influyen en la capacidad de ejercicio y la morbimortalidad a corto y largo plazo. Aunque las intervenciones hemodinámicas para reducir la sobrecarga de volumen producida por la IT o la IPparece que aportan una mejora hemodinámica y dela clase funcional, continúan produciéndose cambios en cuanto al momento adecuado para practicarla cirugía valvular. En estos pacientes, lo más adecuado es que cardiólogos expertos en cardiopatíascongénitas en el adulto realicen evaluación y seguimiento en centros experimentados. Los nuevos tratamientos como los DAI y la TRC parecen prometedores en los casos de insuficiencia del VDsistémico, aunque son necesarios más estudios alrespecto.
ABREVIATURAS
CC: cardiopatía congénita.
EP: estenosis pulmonar.
IP: insuficiencia pulmonar.
IT: insuficiencia tricuspídea.
TSVD: tracto de salida ventricular derecho.
VD: ventrículo derecho.
VI: ventrículo izquierdo.
VT: válvula tricúspide.
Conflicto de intereses: El Dr. Rafael Alonso-González ha recibido una beca de investigación de la Fundación Alfonso Martín Escudero, Madrid, España.
El Profesor Gatzoulis y el Royal Brompton Adult Congenital Heart DiseaseCentre and Centre for Pulmonary Hypertension han contado con el apoyode la British Heart Foundation.
Full English text available from: www.revespcardiol.org
Correspondencia: Prof. M.A. Gatzoulis.
Adult Congenital Heart Centre and Centre for Pulmonary Hypertension.Royal Brompton Hospital.
Sydney Street. SW3 6NP, London. Reino Unido.
Correo electrónico: m.gatzoulis@rbht.nhs.uk