Keywords
INTRODUCTION
For over a thousand years, the world's view ofthe pulmonary circulation hewed to the teachingsof Galen, who believed that blood was produced inthe liver, then delivered by the right ventricle (RV)to the tissues and organs where it was consumed. InGalen's view, blood "seeped" into the left ventricle (LV) directly from the RV via invisible pores in theinterventricular septum. While it may now seem selfevident that this is impossible, Galen viewed bloodmovement as a low volume ebb and flow.1
In the 13th century, Ibn al-Nafis of Syria rejectedGalen's description and speculated that bloodfrom the RV reached the LV via the lungs. Whilehe deserves credit for the first accurate descriptionof the pulmonary circulation, his works were lostand largely forgotten until quite recently, and itdoes not seem likely that they influenced theunderstanding of circulatory physiology in thewestern world.2
The first detailed description of the RV andpulmonary circulation to receive significant attentionin the western world appeared near the beginning ofthe 16th century in the midst of a religious discourseby Michael Servetus of Spain. In this work (for whichServetus was later burned at the stake, althoughpresumably for the heretical nature of its religiouscontent, rather than primarily because of his viewson circulatory physiology), Servetus wrote:
[The vital spirit] is generated in the lungs from amixture of inspired air with elaborated, subtle bloodwhich the right ventricle of the heart communicatedto the left. However, this communication is made notthrough the middle wall of the heart, as is commonlybelieved, but by a very ingenious arrangement the subtleblood is urged forward by a long course through thelungs; it is elaborated by the lungs, becomes reddish-yellow and is poured from the pulmonary artery intothe pulmonary vein.3
This model, based strictly on structuralobservations rather than on any experimentalmeasurements, was a dramatic departure fromGalen, but like Galen before him, Servetus assumedblood was continuously produced and consumedrather than re-circulated.3
Fifty years later, William Harvey would developthe first experimentally based model of thecirculation. Despite not being the first to describethe pulmonary circulation, Harvey is considered thefather of modern physiology because he was the firstto perform detailed measurements and calculations1 that allowed him to deduce the existence of blood recirculation, and he demonstrated the pulmonaryblood flow experimentally.4
Over the next 400 years, the importance of the RVwould be debated, with some investigators opiningwell into the 20th century that the RV served nopurpose other than to provide capacitance to thepulmonary circulation.5,6 In large part because of these early investigations, right heart failurewas believed to be a problem mainly confined toidiopathic pulmonary hypertension and congenitalheart disease, where it is a common cause ofdeath. However, it is now known that pulmonaryhypertension (PH) and right heart failure, far frombeing rare, complicate numerous other diseaseprocesses: RV failure is one of the most powerfulpredictors of mortality in left heart failure,7 rightheart failure is the proximate cause of death in mostof the 50 000 fatal cases of pulmonary embolism inthe United States each year,8 and by some estimates,two to six in 1000 people with chronic lung diseasewill develop right heart failure, for several tens ofthousands of new cases a year.9
This review will explore how the interaction of thepulmonary circulation and RV contribute to theirimpact on health and disease.
FETAL AND NEONATAL PULMONARYCIRCULATION AND RIGHT VENTRICLEDEVELOPMENT
By the 3rd week of human gestation, passivediffusion of oxygen into the developing embryobecomes insufficient to support metabolism, bloodhas formed, and the primitive heart tube hasbegun beating; by the end of the 4th week, activecirculation begins. Distinct components of thepulmonary and systemic circulation emerge fromfolding and twisting of the heart tube between the3rd and 5th weeks of gestation, under control of acomplex signaling network that includes the retinoicacid and neuregulin pathways. Soon after, the RVand pulmonary circulation begin to separate fromthe LV and systemic circulation by formation of theinterventricular septum from the endocardial cushion,and the valves develop. At birth, full septation of theinteratrial septum is normally complete, with onlythe foramen ovale remaining as a potential shuntbetween the right and left atria.10-12
In the embryo and fetus, the RV is the dominantchamber, accounting for about 60% of total cardiacoutput. Because the embryo receives oxygen andnutrients from the placenta, only 15%-25% of totalcardiac output enters the lungs. The remainderof right sided cardiac output is diverted to thesystemic circulation via the foramen ovale to theleft atrium and via the ductus arteriosis from thepulmonary artery to the aorta. Between 40%-60% of descending aortic flow enters the placenta viathe umbilical artery, then returns via the umbilicalvein to the liver or through the ductus venosus tothe inferior vena cava.13,14
At birth, pulmonary vascular resistance fallsrapidly after expansion and oxygenation of thelungs, and right ventricular cardiac output beginsto flow predominantly through the pulmonaryartery into the lungs. At that point, rising left atrialpressure seals off the one way "flap valve" of theforamen ovale.15 At birth, RV pressures still exceedsystemic pressures, but these begin to fall over thenext few hours to days.16 Shortly thereafter, theductus arteriosus, under control of prostaglandin,begins to close,14 the LV hypertrophies as it takesover the systemic circulation, and the RV atrophies.By 3 weeks of age, pulmonary pressure has normallyfallen below systemic pressure, and by adulthood thenormal RV is incapable of generating more than 40-60 mmHg acutely.17
ADULT PULMONARY ANATOMY AND CIRCULATION
The pulmonary artery consists of a thin, elasticvessel that ramifies to supply the various lobarpulmonary arteries, pulmonary arterioles andalveolar capillaries. Blood exits the alveolar capillariesvia the pulmonary venules, and returns through asystem of pulmonary venules and branches similarin structure to the pulmonary arterial tree to theleft atrium.18-20 Because gas exchange occurs in thin,highly permeable alveolar membranes, pulmonarypressure must be low to avoid pulmonary edemafrom elevated Starling forces.21
Unlike the systemic circulation, where acircumferential layer of smooth muscle cells in themedia of the arterioles clearly regulates resistance,pulmonary arterioles less than 70 microns indiameter appear to have at most incomplete layersof smooth muscle in the media, leading to anassumption in the past that blood flow regulationwas limited to vessels greater than 100 microns indiameter. Nevertheless, studies show that regulationalso occurs at the level of pulmonary micro vesselsbetween 30 and 200 microns.22 Regulation is underthe control of a poorly understood oxygen sensingmechanism that may depend on calcium or voltagegated potassium channels, reactive oxygen species,or other mechanisms,23 as well as on nitric oxide,prostaglandins, endothelin, and catecholamines.24-26
A number of concepts are commonly applied todescribe the resistance to flow in the pulmonarycirculation, and the consequent "afterload" experienced by the RV. The most completedescription is provided by pulmonary inputimpedance. However, full characterization of input impedance is technically demanding, requiringfrequency domain analysis of simultaneouslymeasured pressure and flow.27,28 The derived frequency domain factors are not easily related tocommon clinical concepts, so simplified modelsare generally preferred. The so-called windkesselmodel29 encompasses three major components ofinput impedance: pulmonary vascular resistance,pulmonary arterial compliance, and a dynamiccomponent called inductance.
Pulmonary vascular resistance (PVR) is defined asthe mean pressure drop from the main pulmonaryartery to the left atrium divided by average cardiacoutput, expressed in dynes-second-cm-5. For convenience of calculation, Wood units are morecommonly used, defined as mean pulmonary arterypressure minus mean pulmonary capillary occlusivepressure in mmHg, divided by cardiac output inL/min. One Wood unit is equivalent to 80 dyne-second-cm-5. PVR is primarily determined by smallvessel resistance, although extrinsic compressionor mechanical obstruction of larger arteries (eg,by emboli) may also alter PVR. Pulmonary arterycompliance refers to the elastic properties of thesystem, and is defined as the ratio of a change involume to a change in pressure; pulmonary arterycompliance buffers flow during RV ejection, reducingpulmonary artery pulse pressure. Inductance describesthe dynamic response to changes in flow due to massand inertia of the blood.
In so-called lumped parameter models (includingthe windkessel framework), resistance is modeled byone or more electrical resistors, compliance is modeledby a capacitor, and inductance is modeled as either aninductor or as a resistor in more simplified models.Various combinations of these elements are possible,but three or four element models are most commonlyused in physiologic investigations. Windkessel modelsprovide considerably better estimates of systembehavior than pulmonary vascular resistance alone.
Several other terms are commonly used inphysiologic studies, although inconsistencies in theirdefinition in the literature may lead to confusion.Effective arterial elastance is defined as the ratioof end systolic pressure to stroke volume, althoughclassically the term "elastance" is simply the reciprocalof compliance. This simple measure lumps togetherstatic and dynamic components of impedance andperforms reasonably well in experimental studies.30,31 While best characterized for use in modeling thesystemic circulation, it has been successfully appliedto the pulmonary circulation as well.32
PULMONARY HYPERTENSION
Table 133,34 shows typical pressures and resistancesof the pulmonary and systemic circulations. Under normal conditions, PVR is 1/20 of systemic vascularresistance, and mean pulmonary artery pressure maynot be much higher than central venous pressure.Because a 5 mm Hg pressure gradient across thepulmonary circulation is sufficient to maintain anormal cardiac output under conditions of normalPVR and normal LV filling pressures, minimalRV contractile function is normally necessary tomaintain cardiac output, permitting congenital heartdisease repair procedures such as the Fontan, wherethe RV is excluded from the pulmonary circulationentirely.35

In contrast to the focus on pulmonary vascularimpedance taken by experimental physiologists,clinicians largely focus on pulmonary arterialpressure as the important operative concept,defining pulmonary hypertension (PH) as a meanpulmonary artery pressure greater than 25 mm Hg,or a peak pressure greater than 35 mm Hg. However,pulmonary artery pressure rises with age, the rangeof normal is wide,36 and pulmonary artery pressureis a function of pulmonary vascular resistance,cardiac output, and pulmonary vein outflowpressure. Thus, a focus on pulmonary arterypressure alone obscures the etiology and potentialtherapeutic options for PH.
In the past, PH was conceptually divided into acuteand chronic, and primary and secondary. Becausethese terms did not provide insight into etiology orpotential therapy, the World Health Organizationdeveloped a new classification system summarizedin Table 2.37,38

Group I PH (commonly referred to as pulmonary arterial hypertension) is defined as PH arising fromprimary abnormalities of pulmonary vasculatureanatomy or function. It includes idiopathicpulmonary arterial hypertension (previously knownas "primary pulmonary hypertension," a term nowabandoned by PH specialists but still in common use).Group I PH is most commonly due to abnormalitiesin the vascular wall of the pulmonary arterioles,although it also includes pulmonary veno-occlusive disease. The exact underlying pathologic changes(reviewed in detail elsewhere)24,39 vary somewhat depending on the etiology, but in most cases thereappears to be a mechanical obstruction to flow anda reduced responsiveness to vasodilators, accountingfor the common term fixed PH (although this issomething of a misnomer since the disease processcan be modified by various therapies). Despitethe comparative rarity of idiopathic PH (a fewthousand new cases per year worldwide), it receivesa disproportionate share of attention from PHspecialists.
Group II PH (commonly referred to as pulmonary venous hypertension) is defined as PH due toretrograde transmission of abnormally elevatedpulmonary vein pressures from a variety of causes.Group II PH is largely secondary to abnormalitiesof left sided heart anatomy or function, eg, LVsystolic or diastolic dysfunction, mitral and aorticvalve disease, and is essentially a passive processwith respect to the pulmonary vasculature. GroupII PH may theoretically be "reversed" by correctionof the underlying pathologic process that led toelevated pulmonary vein pressure. However, inmany conditions (eg, mitral stenosis) increases inpulmonary arteriole resistance may develop overtime and become irreversible, presumably due tosimilar mechanisms as in some forms of Group IPH. Although Group II PH is very common (andindeed, some authors claim that the most commoncause of right heart failure is left heart failure),it is uncertain how much right heart failure inGroup II actually contributes to mortality versussimply being a marker for more advanced leftheart disease.
Group III and Group IV PH are due toalterations in pre-capillary pulmonary arterioles,but in Group III these alterations are secondaryto lung disease or hypoxemia and can to someextent be considered a normal, physiologicresponse to external stimuli. Hypoxic pulmonaryvasoconstriction, which normally matches pulmonary ventilation to pulmonary perfusion,may become pathologic when too many segmentsof lung become hypoxic and PVR rises too far. PHdue to Group III processes may be "reversed" byvasodilators such as calcium channel blockers, directpulmonary vasodilators such as nitroprusside, andinhaled agents such as nitric oxide, but over timeabnormalities of the pulmonary vascular systemmay develop and become essentially permanentas in Group I PH. Moreover, reversal of hypoxicpulmonary vasoconstriction through extrinsicmeans carries the potential to worsen hypoxemiathrough increased ventilation perfusion mismatch.Group III PH likely accounts for many more PHcases than all other groups combined, althoughseverity of Group III PH tends to be somewhatlower than in Group I or Group IV.9
Group IV PH is due to mechanical obstructionor pulmonary arteries or arterioles secondary topulmonary emboli (whether chronic or acute) andtumor emboli. Global statistics are not readilyavailable for this group, but in the United States thereare likely more than 600 000 pulmonary embolismcases causing more than 60 000 deaths per year.8
Group V PH is a catchall for processes that are eitherentirely unknown or that do not fit into one of theother categories.
ADULT RIGHT VENTRICLE ANATOMY
The anatomy of the RV has been reviewed indetail by Ho et al.40 Conceptually, the RV may bedivided into an inflow tract (beginning with thetricuspid annulus), an apical region, and an RV outflow tract (terminating in the pulmonic valve).The RV free wall constitutes the anterior border ofthe RV and consists of a relatively thin crescent ofmuscle, lying anterior to the LV and interventricularseptum. The RV is normally less than 1-3 mm inthickness, in comparison with the 10 mm thick leftventricular free wall, and comprises roughly 1/6thof the total mass of the heart.17 Functionally, theinterventricular septum constitutes the other halfof the RV. Spiral muscle bundles form a contiguousband-like structure functionally linking the RV andthe LV, likely resulting in transmission of contractileforce directly from the LV to the RV.17,41 Short axis cross sections through the heart from apex to basevary from a roughly triangular contour at the apexto a crescentic appearance at the base. This complexshape accounts for the difficulty in assessing RV sizeand function based on two dimensional imagingtechniques,40 and also accounts for the dramaticchanges in RV size and shape that occur with varyingloading conditions.
RIGHT VENTRICLE CORONARY CIRCULATION
In humans, the RV is largely perfused fromthe right coronary artery. In the LV, myocardialperfusion occurs predominantly in diastole whenintramyocardial tissue pressure falls below aorticroot pressure. Under normal loading conditions,RV intramyocardial tissue pressure remains belowaortic root pressure throughout the cardiac cycle,permitting continuous coronary flow, but in severeRV pressure overload the RV coronary perfusionpattern begins to approximate that of the LV.42
NORMAL RIGHT VENTRICLE CONTRACTION
In the LV, development of ventricular pressureand ejection of blood is due to a concentriccontraction of the LV free wall and septum, alongwith a twisting or "wringing" motion of the heart.In contrast, ejection of blood by the RV proceedswith a sequential contraction beginning in the inflowtract, and moving in a wave toward the outflowtract.17,43 Normal ejection from the RV is a functionof both a reduction in RV free wall surface area anda reduction in RV free wall septal distance.44,45
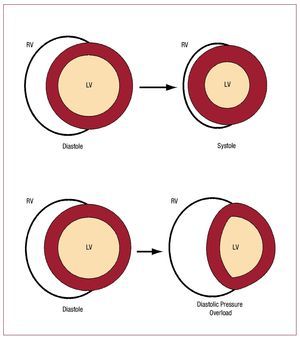
Figure 146 schematizes how the RV and the LVeject blood (neglecting any twisting motion ofventricular motion). Since surface area of a cylinder isproportional to its radius, and volume is proportionalto the square of the radius, ejection fraction in theLV is roughly proportional to the square of thechange in endocardial surface area. In contrast, because of the greater surface area to volume ratioof the RV, a greater ejection fraction is producedby a smaller change in surface area than would berequired in the LV. The bellows-like arrangement ofthe RV not only allows large changes in RV volumewith small changes in RV free wall surface area, butalso helps buffer respiratory changes in RV output47 without necessitating altered contractile function ona breath-to-breath basis.

Figure 1. Illustration of shape changes inthe heart during contraction. The circularcross section LV contracts by a uniformreduction in endocardial surface area,maintaining a nearly constant relationshipbetween volume and surface area. Thecrescentic RV flattens in systole, leadingto a large volume change with minimalchange in RV free wall area. During severepressure overload, the interventricularseptum shifts, increasing RV diastolicvolume with little increase in RV freewall surface area. Without an increasein surface area, the RV cannot recruitadditional function via the Frank-Starlingmechanism. At the same time, there isa reduction in LV end-diastolic volumeand surface area, resulting in impairedLV pump function. (Reproduced fromGreyson CR. Crit Care Med. 2008;36:S57-65. Copyright 2008, with permissionfrom Lippincott, Williams & Wilkins.46). LV indicates left ventricle; RV, right ventricle.
While the series configuration of the pulmonaryand systemic circulation require average RV andLV stroke volume to be the same (in the absenceof intracardiac shunts), RV end-diastolic volume isnormally somewhat greater than LV end-diastolicvolume, while ejection fraction is smaller. Asafterload increases, RV end diastolic volume riseswhile ejection fraction falls.48
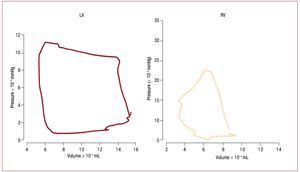
The dynamics of the contracting heart arecommonly analyzed in the context of pressure-dimension plots such as in figure 2.49 The square shape of the LV pressure-volume loop suggestsdefining isovolumic (constant volume) contractionand relaxation phases, and simplifies identificationof end-systole and aortic valve closure, which occurclose to the inflection point of the ejection phase.Suga and Sagawa developed the methodology fordescribing left ventricular function in terms of a "time varying elastance", where elastance is equalto the slope of the pressure volume relation atspecific ("isochronal") times in the cardiac cycle.50 Experimentally, it has been found that the maximumslope of the pressure-volume relation of the LV,called Emax, usually occurs at end-systole and isdirectly related to the contractile state of the LV.49 In most cases it is essentially equivalent to the endsystolic elastance, Ees. In contrast, ejection of bloodthrough the pulmonary valve may continue evenwhen RV pressure is falling due to momentum ofthe blood into the low input impedance pulmonarycircuit. This late ejection, or "hangout period",51 makes identification of "end-systole" problematicin the RV, and contributes to the more triangularshape of the RV pressure-volume loop. The resultis that pressure-volume loops are more difficult to interpret in the RV than in the LV,49,52 and the endsystolic pressure volume relation is not necessarilythe preferred method for assessing RV contractilefunction.53

Figure. 2. Comparison of pressure volume loops obtained in humans with micromanometer catheters and ventriculography in the LV (left) and RV (right). LVpressure volume loops are nearly square, simplifying identification of isovolumic contraction and relaxation phases. In contrast, the RV loop is more triangular,with poorly defined end-systole. (Reproduced from Redington AN. Br Heart J. 1988;59:23-30. Copyright 1988, with permission from BMJ Publishing GroupLtd.49). LV indicates left ventricle; RV, right ventricle.
VENTRICULAR-VASCULAR COUPLING
In any mechanical or electrical system, transmissionof power from one part of the system to another ismaximized when the output impedance of the powerproducing part and the input impedance of the powerreceiving parts of the system are equal. As describedpreviously, elastance is related to impedance, somaximal power transfer from the ventricle to thevascular system is achieved if ventricular (Emax) and vascular (Ea) elastance are equal.54 However, in thebeating heart, where the mechanical properties arechanging over time, theoretical and experimentalstudies have shown that maximum efficiency of workproduction (ie, stroke work to oxygen consumptionratio) is achieved when the ratio of Emax to Ea is closer to 2.55 Under normal conditions, systemiccirculation ventricular-vascular coupling is foundexperimentally to achieve near maximum efficiency.The RV has a less clearly defined end-systole, and Emax is consequently more difficult to define. Nevertheless,several investigators have found that RV-pulmonaryvascular coupling can be analyzed in a similar way,and that, if end-systole is suitably defined, couplingis also nearly optimal under normal conditions (ie,the ratio of RV Emax to pulmonary artery elastanceEa is close to 2).56,57
CONTRIBUTION OF RIGHT VENTRICLE TO CARDIAC OUTPUT
In 1943, Starr et al ablated the RV in open chestdogs with "a red hot soldering iron," and foundlittle alteration in systemic circulation or venouspressure. They speculated that the RV free wallserved no other purpose than to provide capacitanceto the pulmonary circulation.5 Other investigatorssubsequently came to similar conclusions,6 andinterest in the RV began to wane.
However, RV pressure development is acombination of interactions among the RV freewall, the interventricular septum, and the LVfree wall,58,59 and the importance of RV free wallcontractile function depends in large part onpulmonary vascular resistance and RV pressure.For example, while right coronary artery (RCA)occlusion and RV free wall contractile dysfunctionmay have little effect on RV pressure development orsystemic hemodynamics under normal conditions,RV ischemia results in systemic hypotensionwhen pulmonary vascular resistance increases.60 Experimental evidence of the importance of RVcontractile function was given increased creditfollowing Cohn's case series of hemodynamicallyunstable isolated RV infarcts, and subsequent reportsthat RV contractile dysfunction in the setting ofmyocardial infarction (MI) resulted in substantiallyincreased morbidity and mortality.61,62
RESPONSE OF RIGHT VENTRICLE TO ACUTELY INCREASED PRESSURE
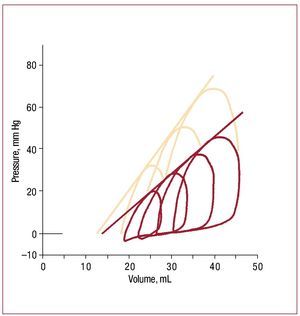
How the heart responds to pressure overloadhas been studied for more than a hundred years,beginning with Otto Frank's efforts to systematizethe study of ventricular function.63 Suga and Sagawaextended and developed these concepts during the1960s and 70s to the point where pressure volumerelations are now routinely discussed in a clinicalcontext.64 Figure 3 shows pressure volume loops inan isolated RV as end-systolic pressure is altered.Each loop represents a single cardiac cycle.52 In broad terms, over a physiologic range, end systolicvolume is roughly inversely proportional to endsystolic pressure, while stroke volume (and hencestroke work) increases with increasing end-diastolicvolume.

Figure 3. Example of RV pressure-volume loops obtained in an isolatedworking dog heart under baseline conditions (red lines) and followinginotropic stimulation with epinephrine (salmon lines). The series of loops ineach case is obtained by varying the outflow resistance. Note that a nearlystraight line can be drawn that is tangent to each loop under a particularcondition; although end-systole is difficult to identify, this line is essentiallyequivalent to the end-systolic pressure volume relation as obtainedin isolated left ventricles, where the slope is a reflection of underlyingcontractile state. (Reproduced from Maughan WL. Circ Res. 1979;44:309-15. Copyright 1979, with permission from Wolters Kluwer Health.52). RV indicates right ventricle.
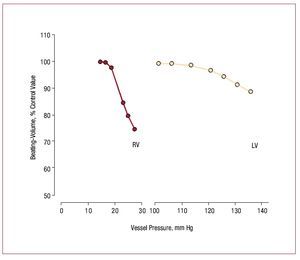
However, in comparison with the LV, the adultRV has very limited capacity to produce elevatedpressure. Those limits are illustrated in Figure 4,which shows that stroke volume as a fraction ofcontrol declines much more quickly in the RV thanin the LV as mean ejection pressure increases.65 The Laplace relation helps explain the RV's morelimited ability to contract against a load. First, thethinner RV free wall experiences a greater rise in walltension with increments in RV pressure. Second,the radius of curvature of the RV increases duringcontraction rather than decreasing as happens in theLV, which means that shape dependent reduction ofstress during contraction due to declining radius ofcurvature does not occur in the RV as it does in the LV.66 There also appear to be biochemical differences,with RV myocardium being more optimized forrapid contraction,67 although whether differencesin myosin heavy chain isoform composition explainthis is uncertain, since RV-LV differences in myosinisoform expression appear to be present in rodents68 but not in dogs.69

Figure 4. Comparison of the effect ofejection pressure on ejection fraction in theRV and the LV. Note that for any incrementin ejection pressure, the decrement inejection fraction is much greater in the RVthan in the LV. (Reproduced from HeartDisease, Braunwald E., ed., by WiedemannHP, Saunders Company, 1997, p. 1606.Copyright 1997, with permission fromElsevier.65). LV indicates left ventricle; RV,right ventricle.
Several mechanisms potentially contribute toincreased contractile function in the setting ofincreased demand. These include the Anrep effect(homeometric auto regulation), the Frank-Starlingmechanism (sometimes referred to as heterometricauto regulation), and catecholamine inducedinotropy.
The Anrep effect is an intrinsic increase incontractile function that occurs in response toincreased afterload in the absence of externalregulatory changes such as catecholaminestimulation. Anrep effect can be demonstrated inisolated muscle strips,69 but its presence in vivo has beencontroversial. Some evidence suggests that Anrepeffect is the primary mechanism for initial adaptationto pressure overload in the RV,70,71 although this maybe more important in neonatal than in adult RVs.
The Frank-Starling mechanism is often viewed asthe primary means by which the heart adapts to anincrease in demand, but shape differences betweenthe RV and the LV alter how the Frank-Starlingmechanism operates. In both RV and LV, an increasein end-systolic pressure is normally accompanied byan increase in both end -systolic and end- diastolicvolume. However, in the RV under normal loadingconditions, much of the increase in volume is due toan increase in RV free wall septal dimension, withmuch less increment in RV free wall surface area. Because the increment in RV free wall area for agiven increment in central venous pressure is small,recruitment via the Frank-Starling mechanismis reduced. Thus, Frank-Starling mechanismplays a smaller role in RV adaptation to increasedafterload at low RV pressures than it does in the LV.At increased afterload, as the RV becomes morecylindrical and other compensatory mechanisms areexhausted, the Frank-Starling mechanism becomesmore important.70,71 Sympathetic stimulation alsoincreases contractile performance in the RV just asin the LV.52
RESPONSE OF RIGHT VENTRICLE TO CHRONICALLY INCREASED PRESSURE
As mentioned previously, RV pressures arenormally higher than systemic pressures in the fetusand neonate, but fall to adult levels in the first fewdays or weeks after birth. However, neonatal RVs cantolerate ongoing PH, and congenital heart diseasepatients tolerate supersystemic pulmonary pressuresfor years with little disability (eg, Eisenmengersyndrome).72 In contrast, once the RV atrophiesand pressures fall, future pressure increases (eventhose that develop over prolonged periods) arepoorly tolerated. The reason for this difference isunknown.
RV hypertrophy may develop in chronic pressureoverload,73 although whether this helps to normalizestress or results in contractile dysfunction isuncertain.74 At the same time, RV dilation maylead to greater recruitment of function through theFrank-Starling relation.
Numerous biochemical alterations have beenreported in various models of chronic RV pressureoverload:75 rodent models of RV pressure overloadexhibit fetal gene program re-expression76,77 and alterations in metabolic, stress-related and structuralproteins,78,79 although large animal models are scarceand not necessarily concordant with findings inrodents.80 Human data obtained from transvenousendocardial biopsies should be interpreted cautiouslybecause alterations in the RV septum, from whichbiopsies are normally obtained, may differ fromalterations in the RV free wall, where much of thestructural alteration is occurring.
RESPONSE OF RIGHT VENTRICLE TO CHRONICALLY INCREASED VOLUME
RV volume overload due to intracardiac shuntsor tricuspid or pulmonic regurgitation is normallywell tolerated, likely because the RV is optimizedto accommodate large changes in volume.Experimentally, chronic RV volume overload doesnot appear to impair RV contractile function,81 and clinically, patients with volume overload due tocongenital heart disease appear to do well for manyyears.82
However, severe RV dilation ultimately impairsfurther volume increase (either due to pericardialconstraint or because of the shared architecture ofthe RV and LV), such that additional incrementsin RV volume recruit minimal increments in RVfree wall surface area; instead, as RV pressure risesfurther, increased RV volume occurs at the expenseof the LV. This is seen most clearly in pulmonaryembolism, where volume loading may adverselyaffect LV function.83
DEVELOPMENT OF RIGHT HEART FAILURE
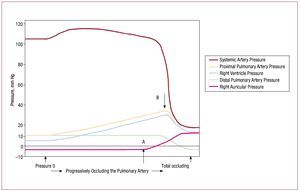
Right heart failure, defined as the inability ofthe RV to generate adequate forward flow withnormal central venous pressure,46 can develop acutely in response to increases in pulmonaryvascular resistance for many reasons, suchas mechanical obstruction of the pulmonaryvasculature by pulmonary emboli, or from reactivevasoconstriction in response to hypoxemia.Experimental data obtained more than 50 years agoshowed that the RV has a very limited capacity tocompensate for such loads. Figure 5 shows the resultof progressively occluding the pulmonary artery in open chest dogs.84 Initially, the rise in pulmonaryartery pressure is well tolerated, with essentiallyunchanged systemic pressure (likely due to Anrepeffect, as discussed previously). At point A, centralvenous pressure begins to rise, recruiting functionvia the Frank-Starling relation; when RV pressurereaches a threshold level at point B, systemicpressure drops abruptly and catastrophically.

Figure 5. Result of progressively occluding the pulmonary artery in an open chest dog. Initially, a progressive rise in pulmonary artery pressure is welltolerated, with essentially unchanged systemic pressure. At point A, central venous pressure begins to rise, permitting recruitment of function via theFrank-Starling relation, until RV pressure reaches a threshold level at point B, at which point systemic pressure drops abruptly and catastrophically.(Reproduced from Guyton AC. Circ Res. 1954;2:326-32. Copyright 1954, with permission from Wolters Kluwer Health.84). RV indicates right ventricle.
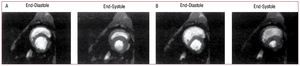
The mechanism of RV failure and hemodynamiccollapse at this point is due to the interaction ofseveral factors. Just prior to hemodynamic collapse,reduced cardiac output may occur consequent tointerventricular interaction. As the RV dilates inresponse to pressure overload, restraint from thepericardium and from shared muscle fiber bundlesof the RV and LV constrain further RV dilation andshift the RV diastolic pressure-volume relation to asteeper portion of the curve, so that further increasesin RV pressure result in less RV free wall stretch andhence less recruitment of function via the Frank-Starling relation. At the same time, interventricularseptal shift impairs LV ejection. This combinationresults in a net decrease in cardiac output. Figure6 shows short axis views of dog hearts subjected toexperimental pulmonary embolism: much of theincrease in RV volume comes at the expense of LVvolume.85

Figure 6. Short axis images from the ventricle of a dog obtained using magnetic resonance imaging in diastole and systole under baseline conditions (A)and following experimental pulmonary embolization (B). Increased RV pressure overload results in RV diastolic dilation and septal shift from the RV to the LV,compromising LV filling and reducing LV output. (Reproduced from Dell'Italia LJ. J Appl Physiol. 1995;78:2320-7. Copyright 1995, with permission from TheAmerican Physiological Society85). LV indicates left ventricle; RV, right ventricle.
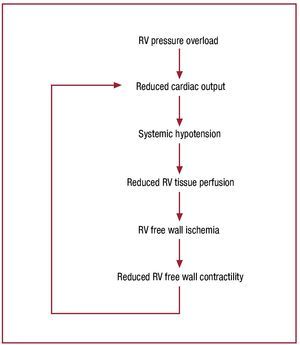
Once cardiac output begins to fall, hemodynamiccollapse progresses rapidly. Figure 746 schematizes thelikely mechanism of hemodynamic collapse: increasedRV pressure overload due to elevated afterload resultsin reduced cardiac output and systemic hypotension;this reduces RV tissue perfusion pressure; once thisdrops below a critical value, RV free wall ischemiadevelops.86,87 RV ischemia causes reduced contractilefunction, compromising the RV's capacity to handlethe elevated RV afterload, further reducing cardiacoutput, and causing a rapidly progressive downwardspiral to hemodynamic collapse.

Figure 7. Proposed schema for the mechanism of abrupt hemodynamicdecompensation in the setting of a severe increase in pulmonary inflowimpedance. Once pulmonary resistance reaches level B in figure 5, RVoutput falls below a critical level, resulting in systemic hypotension,precipitation of RV ischemia, a decline in intrinsic RV contractile function,further decrease in RV output, and a progressive downward spiral tohemodynamic collapse and death. In the absence of some intervention toreduce pulmonary impedance or increase cardiac output, this process isirreversible. Reproduced from Greyson CR. Crit Care Med. 2008;36:S57-65. Copyright 2008, with permission from Lippincott, Williams & Wilkins46). RV indicates right ventricle.
Because hemodynamic collapse in this scenario isabrupt (and frequently irreversible), and right heartfailure does not become manifest by elevated centralvenous pressure until RV compensation is nearlyexhausted, many (if not most) patients who presentwith signs and symptoms of right heart failure arelikely found within a very narrow hemodynamicrange, where central venous pressure has begunto rise but systemic pressure has not yet begun tofall (between points A and B in fig. 5). Recentexperimental evidence suggests that progressiveRV contractile dysfunction unrelated to ischemiadevelops in acute RV pressure overload25,88-90 within this range, and potentially contributes tosudden hemodynamic deterioration in patients whootherwise appear hemodynamically stable.
Several mechanisms may contribute to progressiveRV dysfunction in this setting. Abnormalities of ventricular-vascular coupling can reduce theefficiency of power transmission from the RV to thepulmonary circulation.25 While some investigatorsbelieve that chronic ischemia plays a role inright heart failure, interventions to increase RVcoronary perfusion in models of pressure-overloadinduced failure have not consistently improved RVcontractile function,91 and it is clear that progressive contractile dysfunction can occur in the absence ofany evidence of ischemia at all.25,88-90 Activation of intracellular proteases such as calpain,92 or activation of apoptotic (programmed cell death)pathways93,94 may contribute to dysfunction in thissetting. Calpain inhibition appear to attenuatedysfunction92 and reduce RV apoptosis94 frompressure overload. Calpain may cause contractile dysfunction via degradation of intercellularadhesion proteins such as talin that coordinatecardiac contraction.95,96 A recent report identifies tenascin, an extracellular matrix linking protein, asa possible culprit in chronic heart failure.97 Whetherthe changes in gene expression discussed previouslyare adaptive or maladaptive is unknown.
IMPLICATIONS FOR FUTURE RESEARCH
In theory, progressive contractile dysfunctioncould alter the threshold at which the downwardspiral of hemodynamic collapse begins. Perhapsthis could explain why patients with apparentlystable pulmonary hemodynamics can abruptlydecompensate. Alternatively, RV contractiledysfunction might render a patient more susceptibleto hemodynamic collapse following a subsequentevent that further compromises RV contractilefunction. Improved understanding of the mechanismof RV failure in the setting of PH is likely to increase interest in this neglected disorder, and may facilitatedevelopment of new therapeutic approaches thatgo beyond merely reducing pulmonary vascularimpedance and begin to address the underlyingmechanisms of RV failure.
ABBREVIATIONS
Ea: effective arterial elastance
Emáx: end-systolic elastance
Esf: maximum elastance
LV: left ventricle
PH: pulmonary hypertension
PVR: pulmonary vascular resistance
RV: right ventricle
Supported by the United States Department of Veterans Affairs, and the United States National Heart, Lung and Blood institute grantsR01-HL068606 and R01-HL49444.
Correspondence: C.R. Greyson,
Cardiology 111B, Denver VAMC,
1055 Clermont Street. Denver, CO 80220, USA
E-mail: Clifford.Greyson@UCDenver.edu; Clifford.Greyson@VA.gov