Congenital long QT syndrome (LQTS) is a hereditary disease characterized by abnormal cardiac repolarization (QT interval prolongation) and susceptibility to sudden cardiac death due to torsade de pointes. Its most severe form, Jervell and Lange-Nielsen syndrome (JLNS), is associated with sensorineural deafness and is caused by homozygous or compound heterozygous mutations in 2 possible genes (KCNQ1 and KCNE1). Its prevalence is very low, affecting 1 to 4 million children worldwide (less than 1% of patients with LQTS), and limited numbers of cases have been reported in Spain.1 We present 2 families with JLNS linked to the same previously undescribed variant (KCNQ1 c.604 + 1G>C) found in homozygosity in 1 case and in compound heterozygosity in the other, together with a molecular analysis of the changes caused by this variant.
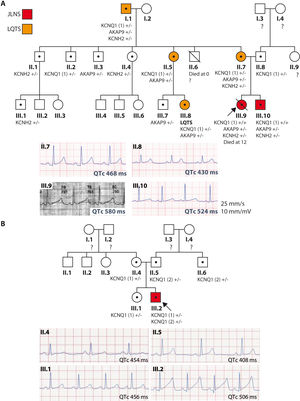
Family 1: the index case (III.9, figure 1A) is a girl with JLNS diagnosed at birth (QTc > 550ms) who was asymptomatic with propranolol treatment and whose first manifestation was sudden cardiac death at 12 years old while swimming. She was a homozygous carrier of the KCNQ1 c.604 + 1G>C variant, as well as heterozygous for the KCNH2 c.38C>A and AKAP9 c.7010A>G variants, none previously described. Her 5-year-old brother (III.10) is a carrier of the same variants, also has JLNS (QTc > 500ms), and is asymptomatic with nadolol and prophylactic defibrillator implantation at 7 years old. The family has a history of consanguinity in distant ancestors, as well as of sudden cardiac death. A family study was conducted according to current recommendations.2 All family members underwent cardiac stress testing to assess their QT interval response to exercise. Their phenotype was considered normal if the baseline QTc interval was < 450ms in male relatives or < 460ms in female relatives and the exercise response was normal. Several family members are heterozygous carriers of the variants and exhibit incomplete penetrance and variable expressivity; none had severe manifestations.
 and resting ECG recordings in lead II (lower panel) of the members of families 1 (A) and 2 (B). Red, JLNS; orange, LQTS; white, normal phenotype; •, carrier; ?, not studied; arrow, proband; KCNQ1 (1), c.604 + 1G>C variant; KCNQ1 (2), c.1513_1514delCA; JLNS, Jervell and Lange-Nielsen syndrome; LQTS, long QT syndrome.")
Family trees and electrocardiographic recordings of the families. Family tree (upper panel) and resting ECG recordings in lead II (lower panel) of the members of families 1 (A) and 2 (B). Red, JLNS; orange, LQTS; white, normal phenotype; •, carrier; ?, not studied; arrow, proband; KCNQ1 (1), c.604 + 1G>C variant; KCNQ1 (2), c.1513_1514delCA; JLNS, Jervell and Lange-Nielsen syndrome; LQTS, long QT syndrome.
Family 2: the index case (III.2, figure 1B) is a newborn who was examined due to severe bradycardia; he had a QTc > 500ms and sensorineural deafness (JLNS). A next-generation sequencing study revealed the heterozygous variant KCNQ1 c.604 + 1G>C, which is associated with the KCNQ1 c.1513_1514delCA variant, previously identified to be pathogenic (ClinVar, ID52985). He was asymptomatic with nadolol treatment. The parents and other relatives are heterozygous carriers of the variant, with variable phenotypic expression concerning the QTc interval, and remain asymptomatic.
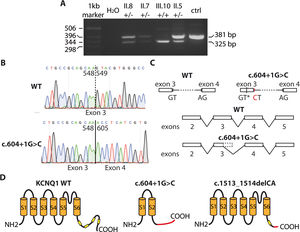
The intronic variant KCNQ1 c.604 + 1G>C is not included in any public database and is of uncertain significance. To determine its effect on the protein (Kv7.1 channel), a molecular study was performed, with the approval of the hospital ethics committee. The patients gave informed consent for the performance and publication of the work. Given its location in the first base of intron 3 to 4, we suspected that the mutation would affect mRNA splicing. For this reason, using RNA extracted from peripheral blood mononuclear cells, RT-PCR was performed to amplify the sequence around the variant. In homozygosity (proband, III.10), the amplified band was smaller than that from a healthy person, whereas heterozygous patients (II.5, II.7, and II.8) showed 2 bands (figure 2A). Sequence analysis of the bands indicated that the c.604 + 1G>C variant caused a loss of the splicing donor site at the start of intron 3 to 4. Consequently, the cell machinery appeared to use a cryptic splicing site located in exon 3, resulting in the loss of a section, as can be seen in figure 2B and in line with the descriptions of intronic mutations in other genes.3 This led to the generation of a truncated and nonfunctional protein (p.Y184Pfs*82; with 264 amino acids vs the 676 in the wildtype channel) (figure 2C). Accordingly, homozygous patients do not generate the IKS current and show a more severe phenotype (JLNS). In the case of family 2, the c.1513_1514delCA variant introduces a change in the reading frame and also produces a truncated Kv7.1 channel (p.Q505Afs*10; figure 2C) with 513 amino acids. This channel lacks a large part of the carboxy terminus, including 3 amphipathic helices (B, C, and D), which would alter the tetramerization of the channel subunits and their trafficking to the plasma membrane.4 Consequently, the index patient also lacked functional Kv7.1 channels and could not generate IKS current. In contrast to this severe condition, it is common to find family members who are heterozygous for KCNQ1 and have a much more benign phenotype. The variants associated with JLNS (typically frameshift or truncation) do not usually exert a dominant-negative effect under heterozygous conditions, as was seen in both families. The additional effects of the other variants of uncertain significance on the phenotype of family 1 is the subject of another investigation, although it is suspected that an additive polygenic effect explains the observed phenotypic heterogeneity.
 and a carrier of the c.604 + 1G>C variant. C: effects of the variant on mRNA splicing. AG, acceptor site; GT, donor site; GT*, cryptic donor site; CT, variant. D: effects of the c.604 + 1G>C and c.1513_1514delCA variants on the structure of Kv7.1 protein. The yellow boxes represent the amphipathic helices. The area in red of the proteins indicates the sequence with an altered reading frame.")
Effects of the variants in KCNQ1. A: RT-PCR–amplified fragments from carriers of the c.604 + 1G>C variant in family 1. B: chromatograms showing sequences from a health person (upper panel) and a carrier of the c.604 + 1G>C variant. C: effects of the variant on mRNA splicing. AG, acceptor site; GT, donor site; GT*, cryptic donor site; CT, variant. D: effects of the c.604 + 1G>C and c.1513_1514delCA variants on the structure of Kv7.1 protein. The yellow boxes represent the amphipathic helices. The area in red of the proteins indicates the sequence with an altered reading frame.
Our opportunity to study 2 families with JLNS caused by a new variant expressed in 2 different forms (homozygous and compound heterozygous) has enhanced our understanding of the spectrum of presentations typically detected in families with JLNS, from asymptomatic carriers to individuals with different degrees of phenotypic expression, and confirms the “multiple hits” theory as a generator of higher clinical expression.5
FUNDINGThe present work was funded by the Spanish Society of Cardiology (Beca Ritmo 17) and the Consejería de Salud de la Junta de Andalucía (PI-0365-2017).
AUTHORS’ CONTRIBUTIONSE. Arana-Rueda and M.R. Pezzotti contributed equally to this work. E. Arana-Rueda and A. Castellano: study conception, data collection and analysis, and manuscript drafting. Remaining authors: data collection and analysis and critical revision of the manuscript.
CONFLICTS OF INTERESTThere are no conflicts of interest.