El síndrome de QT largo (SQTL) congénito es una enfermedad hereditaria caracterizada por una alteración de la repolarización cardiaca (prolongación intervalo QT) y susceptibilidad a la muerte súbita debida a torsade de pointes. La forma más grave de presentación, el síndrome de Jervell y Lange-Nielsen (SJLN), se asocia con sordera neurosensorial y se debe a variantes en homocigosis o heterocigosis compuesta de 2 posibles genes (KCNQ1 y KCNE1) y tiene una prevalencia muy baja, 1-4 millones de niños (menos del 1% de los pacientes con SQTL), y los casos descritos en nuestro medio son anecdóticos1. Se presentan los casos de 2 familias con SJLN debido a una misma variante no descrita previamente (KCNQ1 c.604+1G>C) presentada en homocigosis en un caso y en heterocigosis compuesta en el otro, con un análisis molecular de los cambios provocados por esta variante.
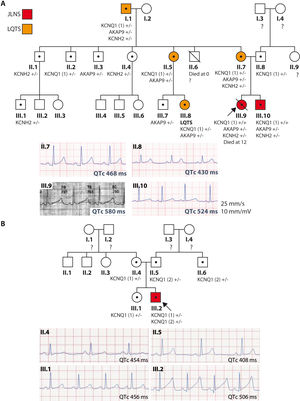
Familia 1: el caso índice (III.9, figura 1A) es una niña con SJLN diagnosticada desde el nacimiento (QTc> 550ms), asintomática en tratamiento con propranolol, cuya primera manifestación fue muerte súbita a los 12 años mientras nadaba. Era portadora de la variante KCNQ1 c.604+1G>C en homocigosis, además de las variantes KCNH2 c.38C>A y AKAP9 c.7010A>G en heterocigosis, ninguna descrita con anterioridad. El hermano (III.10) de 5 años era portador de las mismas variantes, presentaba también SJLN (QTc> 500ms), y permanecía asintomático con nadolol y desfibrilador implantable profiláctico desde los 7 años. En la familia había antecedentes de consanguinidad en antepasados lejanos, con antecedentes de muertes súbitas. Se llevó a cabo un estudio familiar según las recomendaciones actuales2. Se hizo ergometría a todos los familiares para valorar su respuesta del intervalo QTc al esfuerzo. Se consideró un fenotipo normal si el intervalo QTc basal era <450 ms en varones o <460ms en mujeres, y la respuesta al esfuerzo fue normal. Varios miembros de la familia eran portadores de las variantes en heterocigosis y presentaban penetrancia incompleta y expresión variable, en ningún caso con manifestaciones graves.
 y registros de ECG basal en derivación II (panel inferior) de miembros de las familias 1 (A) y 2 (B). Rojo: SJLN; Naranja: SQTL; Blanco: fenotipo normal; •: portador; ?: no estudiado. Flecha: probando. KCNQ1 (1): variante c.604+1G>C; KCNQ1 (2): c.1513_1514delCA. SJLN: síndrome de Jervell y Lange-Nielsen; SQTL: síndrome del QT largo. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Pedigrís y registros electrocardiográficos de las familias. Pedigrí familiar (panel superior) y registros de ECG basal en derivación II (panel inferior) de miembros de las familias 1 (A) y 2 (B). Rojo: SJLN; Naranja: SQTL; Blanco: fenotipo normal; •: portador; ?: no estudiado. Flecha: probando. KCNQ1 (1): variante c.604+1G>C; KCNQ1 (2): c.1513_1514delCA. SJLN: síndrome de Jervell y Lange-Nielsen; SQTL: síndrome del QT largo. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
Familia 2: el caso índice (III.2, figura 1B) es un recién nacido a quien se estudió por bradicardia grave; presentaba un QTc> 500ms y sordera neurosensorial (SJLN). El estudio genético NGS reveló la variante KCNQ1 c.604+1G>C en heterocigosis, asociada con la variante de KCNQ1 c.1513_1514delCA, identificada previamente como patogénica (ClinVar, ID52985). Permanecía asintomático en tratamiento con nadolol. Los padres y otros familiares son portadores heterocigotos de una variante, con expresión fenotípica variable sobre el QTc, y permanecen asintomáticos.
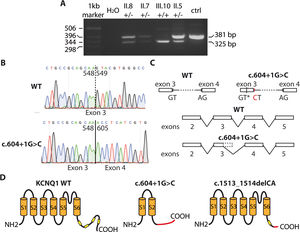
La variante intrónica KCNQ1 c.604+1G>C no está incluida en ninguna base de datos pública, y es de significado incierto. Para conocer su efecto en la proteína (canal Kv7.1), se hizo un estudio molecular, aprobado por el comité ético del hospital. Los pacientes dieron su consentimiento informado para la realización y publicación del proyecto. Al estar localizada en la primera base del intrón 3-4, se sospechó que podría afectar al splicing del ARNm. Por esa razón, a partir de ARN extraído de células mononucleares de sangre periférica, se llevó a cabo una reacción de RT-PCR para amplificar parte de la secuencia alrededor de la variante. En homocigosis (probando, III.10) se amplificó una banda de menor tamaño del de una persona sana, mientras que los pacientes heterocigotos (II.5, II.7 y II.8) mostraron ambas bandas (figura 2A). El análisis de las secuencias de las bandas indicó que la variante c.604+1G>C produce la pérdida del sitio donador de splicing al inicio del intrón 3-4, por lo que la maquinaria celular parece utilizar un sitio críptico de splicing localizado en el exón 3 que conlleva la pérdida de una parte, a la luz de lo que se observa en la figura 2B y en consonancia con lo descrito de mutaciones intrónicas en otros genes3. Como consecuencia, se genera una proteína truncada (p.Y184Pfs*82; con 264 aminoácidos en lugar de 676 del canal silvestre), que no es funcional (figura 2C). Por lo tanto, los pacientes homocigotos no generan la corriente IKs, y muestran un fenotipo más grave (SJLN). En el caso de la familia 2, la variante c.1513_1514delCA introduce un cambio en el marco de lectura y produce también un canal Kv7.1 truncado (p.Q505Afs*10; figura 2C) con 513 aminoácidos. En él faltaría gran parte del extremo carboxilo, incluidas 3 hélices anfipáticas (B, C y D), por lo que se alteraría la tetramerización de las subunidades del canal y su tráfico hacia la membrana plasmática4. Por lo tanto, el paciente índice tampoco posee canales Kv7.1 funcionales y no puede generar la corriente IKs. En contraste con esta afección tan grave, es habitual encontrar que los familiares heterocigotos en KCNQ1 presentan un fenotipo mucho más benigno. Se ha señalado que las variantes asociadas con SJLN (habitualmente frameshift o truncamiento) no suelen ejercer un efecto dominante negativo al presentarse en heterocigosis, como se ha apreciado en ambas familias. El efecto adicional de las otras variantes de significado incierto en el fenotipo de la familia 1 es objeto de otro estudio de investigación, aunque se sospecha un efecto poligénico aditivo como explicación de la heterogeneidad fenotípica observada.
 y una portadora de la variante c.604+1G>C. C: efectos de la variante sobre el splicing del ARNm. AG: sitio acepador; GT: sitio donador; GT*: sitio donador críptico; CT: variante. D: efecto de las variantes c.604+1G>C y c.1513_1514delCA en la estructura de la proteína Kv7.1. Las cajas amarillas representan las hélices anfipáticas. La zona en rojo de las proteínas indica la secuencia en que cambia el marco de lectura. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Efecto de las variantes en KCNQ1. A: fragmentos amplificados en las reacciones de RT-PCR de portadores de la variante c.604+1G>C en la familia 1. B: cromatogramas que muestran las secuencias de una persona sana (panel superior) y una portadora de la variante c.604+1G>C. C: efectos de la variante sobre el splicing del ARNm. AG: sitio acepador; GT: sitio donador; GT*: sitio donador críptico; CT: variante. D: efecto de las variantes c.604+1G>C y c.1513_1514delCA en la estructura de la proteína Kv7.1. Las cajas amarillas representan las hélices anfipáticas. La zona en rojo de las proteínas indica la secuencia en que cambia el marco de lectura. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
Haber podido estudiar a 2 familias con SJLN debido a una nueva variante expresada de 2 formas distintas (homocigosis y heterocigosis compuesta) permite comprender el espectro de presentaciones habitualmente detectadas en familias con SJLN, desde portadores asintomáticos a diferentes grados de expresión fenotípica, y confirman la teoría del «múltiple impacto» como generadora de una mayor expresión clínica5.
FINANCIACIÓNEl presente trabajo ha sido financiado por la Sociedad Española de Cardiología (Beca Ritmo 17) y la Consejería de Salud de la Junta de Andalucía (PI-0365-2017).
CONTRIBUCIÓN DE LOS AUTORESE. Arana-Rueda y M.R. Pezzotti han contribuido de igual manera al trabajo. E. Arana-Rueda y A. Castellano: concepción del estudio, recogida de datos, análisis y redacción del manuscrito. Resto de autores: recogida de datos, análisis y revisión crítica del manuscrito.
CONFLICTO DE INTERESESNo existen conflictos de intereses.