Various specific diseases can lead to dilated cardiomyopathy, but the cause cannot be identified in many cases and the condition is considered idiopathic. Differentiation between idiopathic and secondary forms is essential, as some are potentially reversible.
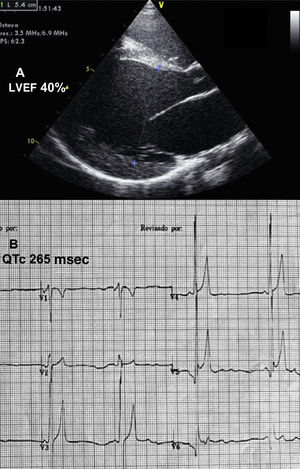
We present the case of an 11-year-old girl, adopted at 1 year of age from Kazakhstan. At 2 years, she was hospitalized in the intensive care unit of another hospital for respiratory distress; a diagnosis of dilated cardiomyopathy secondary to possible myocarditis was established. She additionally had short stature (Z score, –3). The child was treated with angiotensin-converting enzyme inhibitors, diuretics, digitalis, and beta-blockers, and was referred to our unit at 10 years of age because of functional worsening. Echocardiography showed severe left ventricular dilation with an end-diastolic diameter of 54mm, (Z score,+6) and a left ventricular ejection fraction of 40% (Figure 1A). There was marked myocardial trabeculation, which did not meet the criteria of noncompaction, and moderate-severe mitral regurgitation. Magnetic resonance imaging confirmed the echocardiography findings and showed pathological subepicardial gadolinium uptake in the anterolateral region. The electrocardiogram depicted severe sinus bradycardia with abnormal, sharply peaked T waves, high voltage in the middle precordial leads, and an extremely short QT interval (QTc, 265 ms) (Figure 1B). On 24-hour Holter monitoring, there was no evidence of arrhythmia.
 and electrocardiography (B) before treatment. LVEF, left ventricular ejection fraction.")
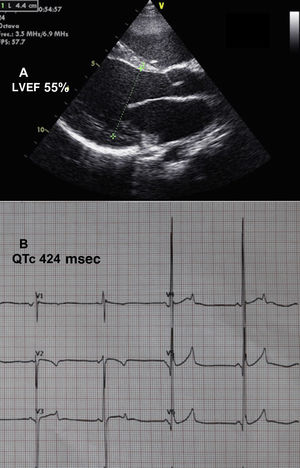
Because of the unusual association of short QT syndrome (usually a purely electrical disease) with cardiomyopathy, we sought a common etiology that could explain the coexistence of the 2 conditions. A literature search for causes of short QT interval found (among others) primary carnitine deficiency (PCD) and other abnormalities related to beta-oxidation of fatty acids. The patient underwent metabolic study, and empirical treatment was started with levocarnitine. Blood ammonia concentration was 365 μmol/L (normal values, 10-60μmol/L), creatine kinase, 260 U/L (normal values, 0-145), and carnitine, 1.3 μmol/L (diagnostic,<5; normal values, 14-69.7). The carnitine dose was gradually increased to 200 mg/kg/d, adjusted according to blood concentrations. Following 6 months of treatment, echocardiography showed a decrease in left ventricular dilation, an improvement in contractility (Figure 2A), and only slight residual mitral regurgitation. The QT interval returned to normal (Figure 2B). At the time of writing, the patient is asymptomatic and heart failure drugs have been discontinued.
 and electrocardiography (B) after treatment. LVEF, left ventricular ejection fraction.")
Genetic study using massive, next-generation sequencing (NGS) detected the c.865C>T variant (p.Arg289*), a pathogenic truncation in the SLC22A5 gene, present in simple heterozygosity. This variant was described previously in combination with a second pathogenic variant in another patient with PCD.1 Analysis of a fibroblast sample showed a clear decrease in intracellular carnitine transport compared with controls, thereby confirming the suspected diagnosis of PCD.
Primary carnitine deficiency is a rare autosomal recessive genetic disease affecting 1:40 000-1:120 000 individuals.2 It is caused by the presence of 2 mutated alleles (either in homozygosity or 2 mutations in compound heterozygosity) in SLC22A5, which codes for the OCTN2 transporter, responsible for intracellular carnitine transport. Carnitine is an essential cofactor that enables long-chain fatty acids to pass through the inner mitochondrial membrane for beta-oxidation. Fatty acids that go unused accumulate in affected tissues. This disease has a wide spectrum of clinical manifestations, such as myopathy, hepatomegaly, hyperammonemia, recurrent episodes of hypoglycemia, and hypertrophic or dilated cardiomyopathy, which may be the only manifestation.2,3 This condition is included in the neonatal screening programs of some autonomous regions of Spain.
In the case presented, although a second variant was not found, fibroblast study confirmed the suspected diagnosis of PCD. We do not know whether unusual genetic abnormalities undetectable by NGS might be present, which could affect the protein (deep intronic variants in promoter regions or variants that affect splicing); functional studies would be needed to confirm this hypothesis.
As to the electrocardiographic characteristics of the condition, abnormalities similar to those secondary to hyperpotassemia were described decades ago in PCD: abnormal, sharply peaked T waves in the middle precordial leads.4 Other authors have reported cardiomyopathy and short QT interval in PCD patients.3,5 In a murine animal model treated with Mildronate (which induces carnitine deficiency), Roussel et al.5 reproduced the DPO phenotype, with the development of cardiomyopathy (hypertrophic) and short QT interval. Some authors have indicated that the predisposition to sudden cardiac death in patients with fatty acid beta-oxidation defects such as PCD results from arrhythmias caused by electrical repolarization abnormalities secondary to ion channel dysfunction. Ferro et al.6 showed that high concentrations of long-chain acylcarnitines change the Ikr potassium current.
The case presented leads to 2 conclusions. First, it is essential to rule out potentially treatable etiologies in all patients with dilated cardiomyopathy, and particularly in children. In the case of a hereditary disease, other family members can be assessed. Second, DPC should be suspected in all patients with short QT and dilated cardiomyopathy. If the condition is diagnosed in time, it can be treated and the cardiac involvement completely reversed.