
Pulmonary hypertension is defined as a mean pulmonary artery pressure ≥ 25mmHg. The pulmonary arterial hypertension (PAH) subtype of this condition is characterized by a pulmonary capillary pressure ≤ 15mmHg and an increased pulmonary vascular resistance (≥3 WU)1; untreated PAH has a poor prognosis due to right ventricular failure. Its causes include idiopathic PAH (IPAH), heritable PAH (HPAH), drug- or toxin-induced PAH, and PAH related to other causes such as congenital heart disease (PAH-CHD). It is called HPAH upon identification of a familial pattern or pathogenic mutation; BMPR2 is the most frequently associated gene, found in up to 75% of patients with HPAH and 25% of those with IPAH.1–3 Mutations in this gene can also favor the development of PAH in some congenital heart diseases.4
A genetic and family study was performed in 8 patients with PAH followed up in our center between 2013 and 2016. Our objective was to determine if the screening results would enable reclassification of the patients. The index cases were studied using massively parallel sequencing with a panel of 16 PAH-linked genes (ACVRL1, BMPR1B, BMPR2, CAV1, EIF2AK4, ENG, FOXF1, GDF2, KCNA5, KCNK3, NOTCH3, RASA1, SMAD1, SMAD4, SMAD9, and TOPBP1). Variants were considered pathogenic if they had an allelic frequency < 0.01% in public databases and met the established pathogenicity criteria.5 Pedigree charts were created and clinical and genetic screening was offered to first- and second-degree relatives.
The patients’ characteristics and the genetic and family study results are detailed in Table 1. Of the 8 index cases studied, the family study allowed the identification of 2 patients with HPAH through a family history of PAH. The genetic study found pathogenic or probably pathogenic variants related to PAH in 4 patients: 1 of HPAH, 2 of IPAH, and 1 of PAH-CHD (the last 3 were later reclassified as HPAH). Only mutations in the BMRP2 gene were detected, all previously described.
Patients’ Characteristics
| Index case | Sex | Age at baseline, y | Baseline type of PAH | Final type of PAH | Other findings | Vasodilator test | mPAP, mmHg | PVR, WU | Gene | Variant detected | Previous description of the variant | Relatives | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Carrier | With PAH | ||||||||||||||
| M | F | M | F | ||||||||||||
| 1 | F | 36 | Heritable | Heritable | - | Negative | 52 | 9.4 | BMPR2 | p.Cys34Phe | Probably pathogenic | 2 | 0 | 0 | 3 |
| 2 | M | 10 | Associated with congenital heart disease | Associated with congenital heart disease and heritable | Small ostium secundum ASD | Negative | 58 | 12 | BMPR2 | p.Arg491Trp | Pathogenic | 0 | 0 | 0 | 0 |
| 3 | F | 41 | Idiopathic | Heritable | Pulmonary arteriovenous fistula <1 cmPostpartum | Negative | 59 | 28.5 | BMPR2 | p.Trp13* | Probably pathogenic | 0 | 0 | 0 | 0 |
| 4 | F | 35 | Associated with congenital heart disease | Associated with congenital heart disease | Large superior sinus venosus ASD | Negative | 48 | 6.9 | - | - | - | - | - | 0 | 0 |
| 5 | F | 34 | Idiopathic | Idiopathic | - | Positive | 40 | - | - | - | - | - | - | 0 | 0 |
| 6 | F | 12 | Idiopathic | Heritable | - | Negative | 55 | 10.2 | BMPR2 | p.Asn442Thrfs*32 | Pathogenic | 2 | 0 | 0 | 1 |
| 7 | F | 39 | Idiopathic | Idiopathic | Postpartum | Positive | 37 | 12.2 | - | - | - | - | - | 0 | 0 |
| 8 | F | 65 | Heritable | Heritable | Negative | 49 | 9.3 | - | - | - | - | - | 0 | 1 | |
ASD, atrial septal defect; F, female; PAH, pulmonary arterial hypertension; M, male; mPAP: mean pulmonary arterial pressure; PVR, pulmonary vascular resistance.
We genetically screened 14 relatives of the patients with a confirmed mutation, identifying 1 affected female carrier and 4 healthy male carriers (Table 1).
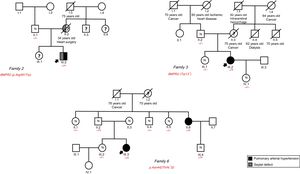
The following mutations were identified: a) case 1: female, 36 years old, classified as HPAH due to the identification of second-degree relatives with PAH; the p.Cys34Phe mutation previously described in another Spanish series6 was found, confirming its cosegregation; b) case 2: male, 10 years old, with a small atrial septal defect and severe PAH-CHD disproportionate to the size of the defect; the p.Arg491Trp mutation was detected and the patient was considered to have dual-etiology PAH–incidental and hereditary congenital heart disease; the boy's mother had a ventricular septal defect, died during corrective surgery, and possibly had PAH; the maternal branch could not be studied because it did not reside in Spain (Figure 1); c) case 3: female, 41 years old, diagnosed with IPAH, with a pulmonary arteriovenous fistula < 1cm and no other typical findings of hereditary hemorrhagic telangiectasia; the cardiac output was greatly reduced, indicating that this finding had no clinical significance; because the p.Trp13* mutation was detected in BMPR2 and no mutation was found in the genes related to hereditary hemorrhagic telangiectasia, she was reclassified as having HPAH; her father was not a carrier, and the presence of the mutation in the maternal branch could not be analyzed (Figure 1); d) case 6: female, 12 years old, with IPAH; because the p.Asn442Thrfs*32 mutation was detected, she was reclassified as having HPAH (Figure 1).

Although the development of PAH has been linked to up to 21 genes,2 our results are consistent with the literature and again show that BMPR2 is the gene most frequently associated with PAH. Penetrance was higher in women and, due to hormonal factors, was often associated with delivery (cases 3 and 7) (Table 1).1–3 In addition, a vasodilator response was more frequent in IPAH6 (cases 5 and 7) (Table 1).
The European Society of Cardiology guidelines recommend the genetic screening of patients with IPAH, HPAH, hereditary hemorrhagic telangiectasia, and pulmonary veno-occlusive disease.1,2 The genetic study of PAH-CHD patients has not been endorsed, although recent evidence shows its usefulness in this context.4 Our series included 2 patients with PAH-CHD; 1 of these patient had a pathogenic mutation in BMPR2, which could indicate its diagnostic value in this subgroup, particularly when the PAH is disproportionate to the degree of the defect or when it develops after defect repair.
In conclusion, our results show that genetic and family screening of PAH enables the identification of hereditary forms and the correct classification of different subtypes, including patients with PAH-CHD with small defects. This approach not only bolsters disease management, but also genetic counseling in families, which can help to avoid transmission of the disease to offspring. For this reason, we believe that screening should be considered in routine clinical practice.
FUNDINGCentro de Investigación Biomédica en Red (CIBERCV), Instituto de Salud Carlos III, CB16/11/00425. Partially supported by an unconditional grant from Actelion Pharmaceuticals.