Keywords
INTRODUCTION
Anomalous origin of a pulmonary artery branch from the aorta is a rare congenital heart disease, in which one of the pulmonary branches arises from the ascending aorta. Aortic arch abnormalities include partial or complete failure in the development of the left sixth arch.1 The condition is included in the group of aortic arch abnormalities and is produced by partial or complete developmental failure of the left sixth arch.1
This defect should be suspected in newborns with heart failure and increased pulmonary flow. Unless surgically repaired, the condition is fatal in most cases due to irreversible pulmonary hypertension, which can be present at very early ages.2,3
We describe the case of a neonate prenatally diagnosed with Fallot's tetralogy, who presented heart failure. Catheterization disclosed Fallot's tetralogy associated with anomalous origin of a pulmonary artery (PA) branch from the left aorta, from the ascending aorta. Surgical correction of the defect was performed and the postoperative course was favorable.
CASE STUDY
Neonate born at term with a prenatal diagnosis of Fallot's tetralogy. The postnatal echocardiography showed a large subaortic ventricular septal defect, aortic arch 50%, pulmonary artery trunk 8 mm, and pulmonary branches 4 mm. The anomalous origin of one pulmonary artery branch from the aorta (Figure B and D) was not visualized, and there was no pulmonary valvular or subvalvular stenosis. In the second week of life, the infant presented progressively worse tachypnea, tachycardia, hepatomegaly, and grade II/VI systolic murmur in the mesocardium. The chest x-ray showed cardiomegaly, pulmonary edema, and an elevated apex of the heart. Therapy with digitalis drugs and diuretics was initiated, with partial improvement.

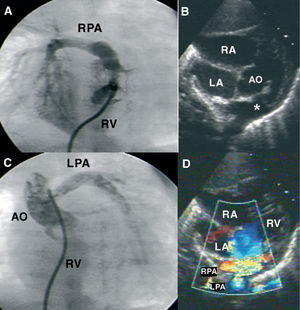
Figure. A: right ventriculography: pulmonary artery (PA) trunk, right PA, and absence of left PA are visualized. B: subcostal view: a ventricular septal defect and pulmonary infundibular stenosis can be seen. Note the false image of both branches emerging from the PA trunk (*). C: ascending aortography; opacification in the left PA, which originates from the lateral side of the aorta, is observed. D: same view with color Doppler. RA indicates right atrium; LA, left atrium; Ao, aorta; RPA, right pulmonary artery; LPA, left pulmonary artery; RV, right ventricle.
Since the clinical symptoms were not consistent with the echocardiographic findings, cardiac catheterization was performed, disclosing good right ventricle (RV) function, an infundibular-valvular gradient of 20 mm Hg, PA trunk with no left PA (Figure A), and pulmonary hypertension due to pulmonary overflow (Qp/Qs 2.4) with normal resistances; both ventricles had the same pressures. The aortography disclosed a right aortic arch and origin of the left PA from the posterolateral wall of the ascending aorta (Figure C). Left ventriculography showed a large subaortic ventricular septal defect.
The microdeletion 22q11 study was positive. At 23 days of life, surgical repair was performed, consisting of resection of the left PA and reimplantation on the PA trunk, along with direct closure of the ascending aorta and subaortic ventricular septal defect with a heterologous pericardium patch. The infundibular muscle and anomalous bands of the RV were resected through an atrial approach. Postoperative echocardiography showed a gradient of 17 mm Hg at the left PA and RV pressures at 40% of systemic pressures. One week later, the patient was discharged.
DISCUSSION
Anomalous origin of a PA branch from the aorta is an extremely rare conotroncal malformation, accounting for only 0.12% of all congenital heart diseases.4 In most cases, the anomalous branch originates on the posterolateral wall of the ascending aorta, near the aortic valve.5,6 In 15%, the origin is distal, near the base of the innominate artery.4 Anomalous origin of the right branch is 5-6 times more frequent than the left.4,6,7
Most articles on anomalous origin of a PA branch from the aorta describe isolated clinical cases except for 2 series,6,7 which review 12 and 16 cases, respectively.
This condition can present alone or in association with other congenital heart defects; the most frequent is persistent ductus arteriosus.3,4,6 The heart disease most frequently associated with left-sided anomalous origin is Fallot's tetraology.8 Association with microdeletion of chromosome 22q11, as occurred in our patient, has only been described in one case, indicating that this anomaly pertains to the group of conotroncal cardiac malformations caused by anomalous development of the neural crest cells.9
Only 9 cases of anomalous origin of the PA from the left aorta associated with Fallot's tetralogy have been reported; the case we describe is the first reported in the Spanish medical literature.
The clinical presentation includes heart failure, heart murmurs, tachypnea, dyspnea, and repeated respiratory distress. Heart failure is the predominant symptom, with this being particularly serious when associated with coarctation or interruption of the aortic arch.10
Two-dimensional color Doppler echocardiography is used for the initial diagnosis, although up to 15% of cases can go undetected.10 In our case, we could not detect the anomalous origin, probably because both branches were very close, as observed in Figure (B and D). The Doppler examination may be suggestive of anomalous origin,11 visualizing systodiastolic flow in the left PA, a phenomenon that we probably did not observe due to the elevated pulmonary vascular resistance of the newborn. Cardiac catheterization continues to be the definitive test for diagnosis, along with providing useful information for surgical planning.
Catheterization shows the systemic pulmonary pressures in the 2 arteries. The abnormally connected lung is perfused at a systemic pressure, whereas the other is exposed to the entire cardiac output from the RV. When the pulmonary resistances drop during the neonate period, the pressure and flow within the anomalous PA increase, causing overcirculation and leading to pulmonary hypertension.10 The development of pulmonary hypertension in the lung normally irrigated by the pulmonary artery may be due to vasoconstrictive circulating substances or crossed neurogenic factors.12,13
Early surgery within the neonatal period is preferable,11 as it would prevent pulmonary occlusive vascular disease that can develop quickly from age 3 months.2,3 The surgery consists of direct anastomosis of the anomalous branch to the pulmonary artery trunk.3,11 It may be necessary to expand the operated area with an autologous pericardium patch or interposition of a homograft.11 Techniques that use autologous tissues to widen and lengthen the anomalous origin of a PA branch from the aorta may be associated with a lower incidence of restenosis than direct anastomosis techniques,14 although the reported series are too small to clearly demonstrate this. At the present time, mortality is virtually nonexistent.
The most frequent postoperative complication is stenosis of the anastomosis (10.6%), which can develop months after surgery.7 Patients with severe restenosis may require surgical angioplasty, balloon dilatation or stent placement.7
Anomalous origin of a PA branch from the aorta is a rare entity in which prompt diagnosis and surgery are essential to prevent irreversible vascular pulmonary disease. Follow-up to detect restenosis, which is frequent, is important.
Correspondence: Dr. F. Prada.
Sección de Cardiología Pediátrica. Hospital Sant Joan de Déu.
Passeig de Sant Joan de Déu, 2. 08950 Esplugues. Barcelona. España.
E-mail: fprada@hsjdbcn.org