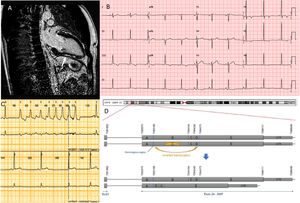
A 35-year-old amateur cyclist with a heavy training routine was referred due to an abnormal electrocardiogram (ECG). The ECG showed inverted T waves in lateral and inferior leads (Figure 1B). He was asymptomatic and had no personal history of any serious medical condition.

A: Cardiac magnetic resonance imaging. Late gadolinium enhancement is signaled with an arrow. B: Electrocardiogram. C: Holter Monitoring showing a nonsustained ventricular tachycardia. D: Schematic representation of the genetic variant. Upper alleles show normal nonrearranged exons. A,B,C,D,E regions denote theoretic portions of the exon that are rearranged in this complex mutation.
The patient's father died suddenly aged 47 years. The episode was classified as secondary to myocardial infarction. No autopsy was performed. His mother was healthy and none of the family members had previously undergone specific cardiological evaluation. He has 3 siblings and a son, all apparently healthy.
Transthoracic echocardiogram was performed showing only mild (12 mm) left ventricular hypertrophy. Cardiac magnetic resonance imaging showed normal cardiac dimensions, wall thickness, and systolic function. Several intramyocardial and subepicardial areas of late gadolinium enhancement (LGE) were detected in the anterior, lateral and inferior left ventricular walls (Figure 1A). Exercise stress single-photon emission computed tomography (SPECT) imaging showed minor unspecific perfusion defects. No arrhythmias were documented during exercise. Coronary angiography was normal. No rhythm abnormalities were detected on 24 h ECG-Holter monitoring.
A genetic test using a next-generation sequencing panel of 173 genes was performed. Sequencing was completed using an Illumina Hiseq platform. Alignment and filtering of variants was performed in a custom in-house pipeline. Variant pathogenicity was classified according to the American College of Medical Genetics and Genomics guidelines.1
The sequence revealed a complex genetic rearrangement in the DSP gene. (NP_004406.2:p.Ala1904Serfs*7/NC_000006.11:g.7583204_7584375delinsGAGCAGTACCAGGTCGGACTAAGCCAATTTTCATGGCCTCATAAATGCCAAGCTTCTGTTTTGTGGTCTCATTGTATATGCCTGCTATGCAGCTTGAACC). The variant is a copy number variant (CNV) that introduces a stop codon. The CNV consist of a large deletion of 906 base pairs (bp) and an insertion (126 bp) of a downstream region. A portion of the inserted material is also inverted (Figure 1D). The variant has not been reported in the literature or in general population databases. Multiple truncating variants in the region have been associated with arrhythmogenic cardiomyopathy (ACM), especially with left ventricular involvement. The variant was considered probably pathogenic.
In a new risk assessment, 3-week Holter ECG monitoring was performed. Three fast, morphologically complex nonsustained ventricular tachycardias were documented. The patient was put on beta-blockers. Counseling about sports practice was given. The patient was advised to limit his exercise programs to leisure-time activities.
Taking into account the presence of LGE, the type of mutation identified, and the presence of ventricular tachycardias, risk was considered high. A subcutaneous defibrillator (ICD) was implanted. Family screening was completed and revealed that the patient's son, and unexpectedly his mother, were carriers of the variant. The mother was studied with transthoracic echocardiogram, ECG, and magnetic resonance imaging. The son was studied with ECG and transthoracic echocardiogram only. To date, there have been no abnormal cardiological findings in either of these patients, suggesting a precipitating role of the patient's heavy sports practice.
Ayala et al.2 published a review gathering 120 cases reported in the literature. In the cohorts reviewed, 15% of carriers had an episode of sudden death and 61% showed some degree of left ventricular involvement. Castelleti et al.3 reviewed the clinical characteristics of patients diagnosed with ACM carrying DSP mutations. In that cohort, 76% of probands showed left ventricular dysfunction and 93% showed some degree of left ventricular involvement. Missense variants were also associated with high arrhythmic risk.
CNVs are seldom reported in association with ACM. This fact is partly a consequence of technology limitations for the detection of CNVs. However, a few cases have been reported and in some cohorts a specific search for these rearrangements has been performed. CNVs might contribute to a 7% to 10% of the identified variants in ACM.4
ACM with left ventricular involvement can often be misdiagnosed as dilated cardiomyopathy (DCM).2 Frequently, patients only show subtle findings with a high risk of ventricular arrhythmias. We currently lack standardized diagnostic criteria for this entity. Diagnosis is commonly established based on genetic findings.
Current guidelines recommend ICD implantation in patients with DCM showing systolic dysfunction and functional class II or higher. The only genetic etiology taken into account is Lamin mutations. In terms of ACM, an expert consensus published in 2019 includes genetic testing in risk stratification. ICD is recommended in phospholamban, filamin C and Lamin mutation carriers when left ventricular ejection fraction is less than 45%. DSP is not mentioned among the high risk genes, although the relationship between left involvement and variants in this gene is mentioned.5 Experts recommend considering an early implant of ICDs in patients carrying DSP truncating variants due to the high incidence of sudden death reported in small cohorts.2
Our case illustrates the usefulness of nontraditional markers of sudden death. Family history in this case was one of the initial clues that later proved to be a confounding factor. The case describes a complex new variant in DSP causing a subtle phenotype but with high arrhythmic risk. Further research into the clinical phenotype associated with DSP truncating variants and their pathophysiological consequences is required.