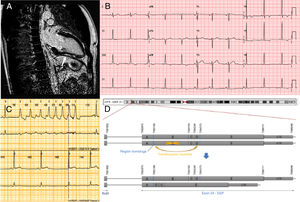
Un ciclista aficionado de 35 años de edad, con una rutina de entrenamiento intensiva, fue derivado a nuestro centro a raíz de un electrocardiograma (ECG) anómalo que mostraba inversión de la onda T en las derivaciones inferolaterales (figura 1B). El paciente se encontraba asintomático y no presentaba antecedentes clínicos relevantes.

A: resonancia magnética cardiaca. El realce tardío de gadolinio se señala con una flecha. B: electrocardiograma. C: registro electrocardiográfico de Holter que muestra taquicardia ventricular no sostenida. D: esquema de la variante genética. Los alelos superiores presentan exones normales sin reordenar. Las regiones A, B, C, D y E ilustran secciones teóricas del exón que aparecen reordenadas en esta mutación compleja. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
El padre del paciente había fallecido de forma súbita a los 47 años, episodio que se consideró secundario a infarto agudo de miocardio, sin que se le practicara la autopsia. Su madre gozaba de buena salud y ningún miembro de la familia se había realizado ningún estudio cardiológico específico. El paciente tenía 3 hermanos y 1 hijo, todos aparentemente sanos.
El ecocardiograma transtorácico reveló únicamente una hipertrofia leve del ventrículo izquierdo (12 mm). La resonancia magnética cardiaca mostró dimensiones cardiacas, grosor parietal y función sistólica normales. Se detectaron áreas de realce tardío de gadolinio (RTG) intramiocárdico y subepicárdico en las paredes anterior, lateral e inferior del ventrículo izquierdo (figura 1A). La tomografía por emisión monofotónica (SPECT) de esfuerzo mostró anomalías menores e inespecíficas de la perfusión. No se presentaron arritmias durante el ejercicio. El resultado de la coronariografía fue normal. Tampoco se detectaron alteraciones del ritmo en el registro electrocardiográfico de Holter de 24 h.
Se efectuó el estudio genético mediante secuenciación de nueva generación de un panel de 173 genes utilizando una plataforma Hiseq de Illumina (Illumina; San Diego, California, Estados Unidos). El alineamiento y filtrado de variantes se llevó a cabo utilizando un algoritmo propio. Se clasificó la patogenicidad de las variantes según las recomendaciones de las guías de práctica clínica del American College of Medical Genetics and Genomics1.
La secuencia mostró un reordenamiento complejo en el gen DSP (NP_004406.2:p.Ala1904Serfs*7/NC_000006.11:g.7583204_7584375delinsGAGCAGTACCAGGTCGGACTAAGCCAATTTTCATGGCCTCATAAATGCCAAGCTTCTGTTTTGTGGTCTCATTGTATATGCCTGCTATGCAGCTTGAACC). La variante corresponde a una variante de número de copias (CNV, por sus siglas en inglés) que introduce además un codón de parada. La CNV consiste en una gran deleción de 906 pares de bases (pb) y la inserción (126 pb) de una región distal. Una parte del material insertado aparece además invertido (figura 1D). No existen reportes de esta variante en publicaciones previas ni se encuentra registrada en bases de datos de población general. Múltiples variantes radicales (tipo truncamiento) en la región se han asociado al desarrollo de miocardiopatía arritmogénica (MCA), en particular, con afección del ventrículo izquierdo. Se estimó que la variante era probablemente patogénica.
Al objeto de evaluar nuevamente el riesgo, se realizó un Holter electrocardiográfico de 3 semanas, donde se evidenciaron 3 taquicardias ventriculares no sostenidas rápidas y de morfología compleja. Se inició tratamiento con bloqueadores beta y se asesoró al paciente sobre la práctica deportiva aconsejándole restringirla a actividad recreativa no extenuante.
Teniendo en cuenta la presencia de RTG, el tipo de mutación identificada y la presencia de taquicardia ventricular, el riesgo se consideró elevado, por lo que se implantó un desfibrilador subcutáneo (DAI). Se completó el cribado de familiares, que puso de manifiesto que tanto el hijo del paciente como de forma sorpresiva su madre eran portadores de la variante. Se realizó el estudio a la madre mediante ecocardiografía transtorácica, ECG y resonancia magnética, mientras que se valoró al hijo solo con ECG y ecocardiografía transtorácica. Hasta la fecha no se han producido hallazgos cardiológicos anómalos en ninguno de los dos, lo cual indica que, en el caso del paciente, la práctica deportiva intensa podría tener un efecto desencadenante.
Ayala et al.2 publicaron una revisión en la que se recogían unos 120 casos con mutaciones truncantes en DSP publicados en la literatura médica. Entre las cohortes examinadas, el 15% de los portadores tuvieron un evento de muerte súbita y el 61% tenía algún grado de afección del ventrículo izquierdo. Castelleti et al.3 revisaron las características clínicas de los pacientes portadores de mutaciones del gen DSP diagnosticados con MCA. De esta cohorte, el 76% de los probandos presentaban disfunción sistólica del ventrículo izquierdo y el 93%, algún grado de afección de este. Además, las variantes sin sentido también se asociaron a un riesgo elevado de arritmias.
Las publicaciones de CNV asociadas a la MCA son poco frecuentes, hecho que es consecuencia en parte de las limitaciones tecnológicas a la hora de detectarlas. Sin embargo, se han reportado algunos casos y en ciertas cohortes se ha efectuado una búsqueda específica de estos reordenamientos. Las CNV contribuyen al 7-10% de las variantes identificadas en la MCA4.
La MCA con afección del ventrículo izquierdo a menudo se diagnostica erróneamente como miocardiopatía dilatada (DCM)2. Con frecuencia los hallazgos son sutiles, si bien los pacientes poseen un alto riesgo de arritmias ventriculares. Actualmente se adolece de criterios diagnósticos estandarizados para esta entidad, y muchas veces ese diagnóstico se establece principalmente basado en los hallazgos genéticos.
Las guías de práctica clínica actuales recomiendan el implante de un DAI en pacientes con MCD que presenten disfunción sistólica en clase funcional II o superior. Las mutaciones del gen de la lamina representan la única etiopatogenia genética considerada. En el contexto de la MCA, la declaración del grupo de consenso de expertos publicada en 2019 incluye el estudio genético para la estratificación del riesgo. Se recomienda el DAI para los portadores de mutaciones del gen del fosfolambán, de la filamina C y de la lamina cuando la fracción de eyección del ventrículo izquierdo sea <45%. No se incluye al DSP entre los genes de alto riesgo, pese a que se haga mención a la relación que se establece entre la afección del ventrículo izquierdo y las variantes de este gen5. Algunos expertos recomiendan el implante precoz de DAI en el caso de pacientes portadores de variantes tipo truncamiento en DSP debido a la elevada incidencia de muerte súbita evidenciada en cohortes pequeñas2.
El presente caso ilustra la utilidad de los marcadores de muerte súbita no convencionales. Los antecedentes familiares del paciente fueron una de las claves iniciales que más tarde resultaron un factor de confusión. Este caso reporta una nueva variante compleja en DSP causante de un fenotipo sutil desde el punto de vista estructural, pero asociado con un elevado riesgo arrítmico. Es necesario continuar investigando en el fenotipo clínico asociado con las variantes tipo truncamiento en DSP y sus consecuencias fisiopatológicas.