La vena cava superior izquierda persistente es la variante del drenaje venoso sistémico más frecuente, con una incidencia del 0,3-0,5% en la población general y del 3-10% en los pacientes con cardiopatías congénitas1. Su presencia se debe a un defecto de la obliteración de la vena cardinal anterior izquierda que drena en la aurícula derecha a través del seno coronario. La dilatación de este constituye el principal signo ecocardiográfico de sospecha. Habitualmente se trata de un hallazgo casual, y en los últimos años es frecuente su diagnóstico durante el periodo prenatal. Lo más habitual es que se presente de forma aislada, pero se describe una mayor incidencia de anomalías cardiacas y extracardiacas asociadas, a la vez que su presencia se relaciona con la aparición de lesiones obstructivas izquierdas2.
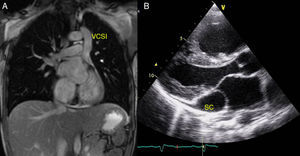
La vena cava superior izquierda persistente sin vena cava superior derecha (figura A) es una anomalía menos habitual causada por la obliteración durante la embriogénesis de la vena cardinal anterior derecha con persistencia de la izquierda. Su incidencia es del 0,09-0,13% en los pacientes con cardiopatía congénita, y en la literatura solo hay publicados casos aislados. La serie más larga consta de 9 casos y una revisión de la literatura3. Se asocia a cardiopatía congénita en el 46% y a trastornos del ritmo en el 36% de los casos3–5.

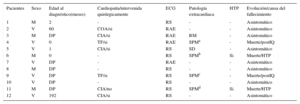
Tras revisar nuestra base de datos desde marzo de 1995 hasta julio de 2015 se han encontrado 150 pacientes pediátricos (0-18 años de edad) con vena cava superior izquierda persistente, y de ellos 12 (8%) con ausencia de vena cava derecha. En la tabla se muestran las características de dichos pacientes. Se trata de 7 varones y 5 mujeres. El diagnóstico se realizó durante un estudio prenatal en 6 casos (50%), posnatal en el contexto de una cardiopatía asociada en 4 (33,3%), y en 2 (16,6%) durante la realización de un estudio ecocardiográfico por otro motivo (soplo inocente y valoración en neonato por sospecha de sepsis). En nuestra serie, 7 pacientes (58%) tenían una cardiopatía asociada: 4 (57%) comunicación interauricular, 2 (28,5%) tetralogía de Fallot y 1 (14,2%) coartación de aorta. Los 5 restantes (42%) tenían un corazón estructuralmente normal. Además, 2 pacientes (uno sin cardiopatía y otro con una comunicación interauricular) presentaron un cuadro de hipertensión pulmonar grave de inicio neonatal. De los 6 pacientes con diagnóstico prenatal, 4 (66,6%) tenían las cavidades izquierdas y el arco aórtico de un tamaño menor que el esperado; 3 de ellos no tenían cardiopatía asociada y presentaron una progresiva normalización posnatal, y el cuarto presentaba una tetralogía de Fallot. En cuanto a trastornos del ritmo, 4 pacientes (33,3%) presentaron un ritmo auricular no sinusal y no se observaron episodios de taquicardia ventricular ni supraventricular. Se hallaron malformaciones extracardiacas en 6 pacientes (50%), que fueron las siguientes: 4 (33,3%) tenían un síndrome polimalformativo (tabla), 1 (8%) presentó hipoacusia y retraso sicomotor no filiado, y otro paciente (8%) tenía síndrome de Down. Los 6 restantes (50%) tenían un fenotipo y un cariotipo normales.
Características de los 12 casos
| Pacientes | Sexo | Edad al diagnóstico(meses) | Cardiopatía/intervenida quirúrgicamente | ECG | Patología extracardiaca | HTP | Evolución/causa del fallecimiento |
|---|---|---|---|---|---|---|---|
| 1 | M | 2 | - | RS | - | - | Asintomático |
| 2 | V | 60 | COA/si | RAE | - | - | Asintomático |
| 3 | M | DP | CIA/si | RAE | RM | - | Asintomático |
| 4 | V | 0 | TF/si | RAE | SPMa | - | Muerte/postIQ |
| 5 | V | 1 | CIA/si | RS | SD | - | Asintomático |
| 6 | M | 0 | - | RS | SPMb | Si | Muerte/HTP |
| 7 | V | DP | - | RAE | - | - | Asintomático |
| 8 | M | DP | - | RS | - | - | Asintomático |
| 9 | V | DP | TF/si | RS | SPMc | - | Muerte/postIQ |
| 10 | V | DP | - | RS | - | - | Asintomático |
| 11 | M | DP | CIA/no | RS | SPMd | Si | Muerte/HTP |
| 12 | V | 192 | CIA/si | RS | - | - | Asintomático |
CIA: comunicación interauricular; COA: coartación de aorta; DP: diagnóstico prenatal; ECG: electrocardiograma; HTP: hipertensión pulmonar; M: mujer; PostIQ, posintervención quirúrgica; RAE: ritmo auricular ectópico; RM: retraso mental; RS: ritmo sinusal; SD, síndrome de Down; SPM: síndrome polimalformativo; TF: tetralogía de Fallot; V: varón.
En la evolución, los 4 pacientes que fallecieron (33%) tenían un síndrome polimalformativo. Dos murieron tras una crisis de hipertensión pulmonar; uno de ellos tenía una comunicación interauricular y falleció antes de la cirugía, y el otro tenía un corazón estructuralmente normal. Los otros 2 pacientes fallecidos presentaban tetralogía de Fallot; uno falleció tras la cirugía correctora y el otro tras una reintervención por insuficiencia pulmonar grave. Los 8 restantes (66,6%) permanecen asintomáticos.
Los 4 pacientes con comunicación interauricular tenían una gran dilatación de las cavidades derechas y 2 de ellos requirieron corrección quirúrgica a una edad más temprana de lo habitual (8 meses y 2 años). Los pacientes con tetralogía de Fallot fueron intervenidos a la edad habitual (antes de los 6 meses).
Se trata de la serie de pacientes con vena cava superior izquierda persistente sin cava derecha con mayor número de casos de todas las publicadas. Como conclusiones podemos decir que se trata de una anomalía muy poco frecuente, que puede diagnosticarse intraútero y que su elevada asociación con malformaciones cardiacas y extracardiacas, así como con síndromes polimalformativos, obliga a realizar un estudio completo en el feto, incluyendo análisis genético. La grave dilatación del seno coronario (tal como muestra la figura B) y la consecuente distorsión del anillo mitral podrían afectar intraútero el llenado del ventrículo izquierdo, lo que justificaría su frecuente asociación con unas cavidades izquierdas pequeñas y con otras lesiones obstructivas izquierdas. En los pacientes con una comunicación interauricular, la compresión del anillo mitral podría favorecer un mayor cortocircuito de izquierda a derecha a través del defecto, lo que justificaría la gran dilatación de las cavidades derechas y la necesidad de una corrección quirúrgica precoz. En nuestra serie, los pacientes sin otras anomalías cursaron de forma asintomática, con normalización posnatal del tamaño de las cavidades izquierdas, por lo que creemos que la alta mortalidad observada en la serie estaría en relación con la presencia de otras malformaciones asociadas.