
Las estrategias de terapia génica incluyen diversos enfoques, como la sustitución y la edición de genes. La sustitución proporciona una copia funcional de un gen alterado y la edición permite corregir una mutación genética preexistente. La terapia génica ya está aprobada para trastornos genéticos como la amaurosis congénita de Leber y la atrofia muscular espinal, y actualmente se estudia su uso en cardiología. En esta revisión se resume el mecanismo de las distintas estrategias de terapia génica, los sistemas de administración disponibles, los principales riesgos relacionados con la terapia génica, los ensayos clínicos en curso y los objetivos futuros, con especial atención a las miocardiopatías.
Palabras clave
La terapia génica humana actúa a través de la transcripción o traducción del material genético transferido o mediante la integración en el genoma del huésped. Estas terapias se administran en forma de ácidos nucleicos, virus o microorganismos modificados genéticamente. El objetivo de esta revisión es analizar el estado actual de las terapias génicas, su aplicación en el tratamiento de las enfermedades cardiovasculares y los retos de futuro, con especial atención a las miocardiopatías.
HISTORIA DE LAS TERAPIAS GÉNICASLa terapia génica humana ha experimentado un rápido desarrollo desde que Theodore Friedmann y Richard Roblin publicaron en 19721 los primeros informes sobre su potencial y los retos que planteaba. Desde entonces, los científicos se han esforzado por elaborar un método seguro, duradero y eficaz para modificar y manipular las células humanas y lograr efectos terapéuticos en diversas enfermedades hereditarias. En 1989 se llevó a cabo el primer ensayo de terapia génica humana aprobado, en que se utilizó un vector retroviral para insertar un gen que codifica la resistencia a la neomicina en linfocitos infiltrantes de tumores humanos para el tratamiento del melanoma metastásico2. Poco menos de 1 año después, otro grupo de investigadores llevó a cabo el primer ensayo de terapia génica aprobado para el tratamiento de enfermedades genéticas hereditarias. En ese estudio, un vector retroviral transfirió una copia funcional del gen de la adenosina desaminasa a linfocitos T explantados de pacientes con inmunodeficiencia combinada grave por deficiencia en adenosina desaminasa. Durante un periodo de 2 años, los 2 pacientes recibieron múltiples infusiones autólogas de células T modificadas3. Estos ensayos clínicos sentaron las bases del potencial de la terapia génica al demostrar la viabilidad, la durabilidad y la seguridad potencial del uso de vectores virales para administrar material genético como aproximación terapéutica.
A pesar de estos estudios de referencia, la muerte de Jesse Gelsinger, de 18 años, en 1999 puso de relieve los peligros de la terapia génica en ese momento. Gelsinger padecía deficiencia parcial de ornitina transcarbamilasa y participó en un ensayo en el que se utilizaron vectores adenovíricos para administrar genes funcionales de ornitina transcarbamilasa. Se inyectaron más de 3,8×1013 partículas adenovíricas en su hígado, lo que provocó un síndrome de liberación de citocinas y dificultad respiratoria aguda que le causaron la muerte4.
Los ensayos clínicos realizados a lo largo de los años noventa y la década siguiente encontraron varios problemas relacionados principalmente con toxicidades y respuestas inmunitarias imprevistas a largo plazo. Un subconjunto de pacientes inscritos en estos ensayos presentó neoplasias leucémicas, ya que el vector retroviral utilizado tenía el potencial de activar protooncogenes5,6. Otros pacientes presentaron amplias respuestas inmunitarias contra los vectores virales y las células recién transducidas que expresan los productos proteicos de la terapia génica7.
En 2012, los premios Nobel Jennifer Doudna y Emmanuelle Charpentier descubrieron un mecanismo de defensa bacteriano denominado CRISPR/Cas9 que podía utilizarse para manipular fácilmente los genomas, lo que sentó las bases para la modificación del genoma8,9. Un mes después, la Agencia Europea del Medicamento aprobó la primera terapia génica (un vector de virus adenoasociados [VAA] que codifica la lipoproteínlipasa, para el tratamiento de la deficiencia de esta)10. En 2017, la Food and Drug Administration (FDA) aprobó en Estados Unidos la primera terapia génica para el tratamiento de la amaurosis congénita de Leber11.
En 2020, la FDA publicó 6 directrices para estandarizar y optimizar el proceso de aprobación de medicamentos de las nuevas terapias génicas12. En respuesta a la pandemia del coronavirus del síndrome respiratorio agudo grave 2 (SARS-CoV-2), la terapia génica logró un gran avance con la aprobación y adopción de 2 vacunas a base de ARN mensajero (ARNm). Estas vacunas utilizaron nuevas nanopartículas de lipoproteínas para suministrar de modo seguro la carga genética en el citoplasma celular13,14. Actualmente se dispone de 27 terapias génicas aprobadas en Estados Unidos, con decenas de ensayos clínicos en curso15.
En el campo de la cardiología, algunos estudios innovadores de terapia génica se llevaron a cabo por primera vez en 1990, y en ellos se utilizó un retrovirus murino para transferir un gen de betagalactosidasa recombinante a la pared de un segmento arterial16. En 1996, Jeffrey Isner utilizó un plásmido que contenía factores de crecimiento vascular endotelial en suspensión en un polímero de hidrogel y recubierto sobre un balón de angioplastia que se aplicó a la arteria poplítea distal para inducir angiogénesis17. Este enfoque sentó las bases de una serie de ensayos clínicos destinados a inyectar plásmidos de ADN desnudo que codifican factores angiogénicos, como el factor de crecimiento endotelial vascular, en las paredes de las arterias coronarias y periféricas, así como en el propio miocardio18–20. Sin embargo, ninguno de estos ensayos tuvo un impacto significativo en los resultados clínicos.
En 2007 se realizó el primer ensayo clínico de terapia génica en pacientes con insuficiencia cardiaca para conocer los efectos de la administración de un gen que expresa SERCA2a a través de un VAA para mejorar la gestión del calcio en los miocardiocitos, donde la Ca2+ ATPasa está desregulada a la baja21. Por desgracia, el tratamiento no logró mejorar los resultados clínicos21,22.
TIPOS DE TERAPIAS GÉNICASEn las enfermedades genéticas, las mutaciones de pérdida de función, por lo general, interruptoras (nonsense), de empalme (splice), de desplazamiento del marco de lectura (frameshift) o de sentido alterado (missense), pueden causar un déficit de producción de proteínas o la liberación de proteínas no funcionales. Las mutaciones por ganancia de función, normalmente a partir de variantes de sentido alterado, determinan la producción de una proteína con actividad aumentada o diferente de la forma de referencia.
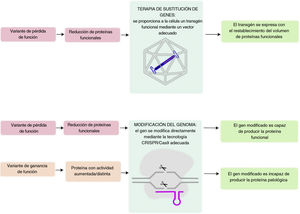
Los abordajes de terapia génica pueden dirigirse a estas alteraciones (figura 1). Es útil dividir las distintas terapias génicas en 2 categorías en función de sus objetivos generales de tratamiento. El silenciamiento génico a través de ARN de interferencia u oligonucleótidos antisentido no se considera estrictamente terapia génica, por lo cual no se trata en este artículo.

Figura central. Las variantes de pérdida de función causan una reducción de proteínas funcionales que podría abordarse mediante terapia de sustitución de genes. Este enfoque implica la introducción de un transgén funcional en la célula afectada, lo que facilita el restablecimiento de la expresión adecuada de las proteínas funcionales. La modificación del genoma comprende varias técnicas que posibilitan la modificación directa del genoma endógeno; en el caso de las variantes con pérdida de función, pueden reestablecer las proteínas funcionales y en el de las variantes con ganancia de función, pueden impedir la expresión de las proteínas patológicas.
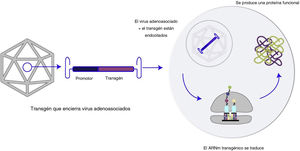
La sustitución de genes tiene como objetivo reemplazar un gen defectuoso por una copia funcional para lograr un efecto terapéutico que atenúe los efectos de una mutación de pérdida de función (figura 2)23. Los productos de sustitución de genes pueden incluir plásmidos de ADN o material genético encerrado en vectores virales, que permiten la transducción de una copia exógena del gen de interés a la célula diana24,25. La terapia génica con plásmidos puede producir un efecto duradero al tiempo que minimiza la inactivación genética del genoma de la célula, lo que lleva aparejado menor potencial oncógeno que las terapias génicas con vectores virales26. Sin embargo, la administración de plásmidos al núcleo es difícil, y es posible que la expresión de proteínas no sea duradera, especialmente en células en división27,28. La terapia génica con vectores virales es capaz de producir una expresión duradera de productos génicos29, pero este enfoque comporta riesgos de toxicidad que se evalúan más adelante en este artículo.
MODIFICACIÓN DEL GENOMA
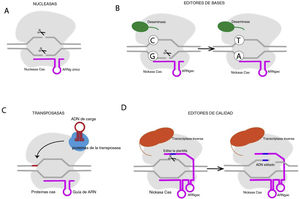
A diferencia de la terapia de sustitución de genes, en la cual se suministra un nuevo gen a través del vector adecuado, la modificación del genoma consiste en modificar la secuencia genómica endógena de la célula y podría utilizarse tanto para mutaciones de pérdida como por ganancia de función. El tipo de modificaciones deseadas se puede obtener a través de la tecnología CRISPR/Cas9 y consisten en: conversión de pares de bases de ADN, eliminación de pares de bases de ADN, inserción de pares de bases de ADN o una combinación de las anteriores (figura 3). CRISPR/Cas9 consta de 2 componentes fundamentales: un ARN guía para que coincida con un gen diana (clustered regularly interspaced short palindromic repeats [CRISPR] o grupos de repeticiones palindrómicas cortas en intervalos regulares) y Cas9, una endonucleasa que determina las roturas de ADN bicatenario (RADNbc). La corrección de las RADNbc puede efectuarse a través de la unión de extremos no homólogos, en la que los extremos de ADN rotos se vuelven a ligar, a menudo con eliminaciones o inserciones de más nucleótidos. Otro mecanismo es la reparación dirigida por homología (homology-directed repair [HDR]), que implica el uso de plantillas de ADN para reparar con precisión el ADN dañado. La HDR solo está activa en células en división30. Las nucleasas CRISPR-Cas se utilizan generalmente para inactivar los genes diana, ya que las variantes «indel» después de la unión de extremos no homólogos generan mutaciones de desplazamiento del marco de lectura que impiden la producción de proteínas funcionales. Los editores de bases contienen una nucleasa CRISPR-Cas incapaz de producir RADNbc (nickasa) y una enzima ADN-desaminasa e incluyen editores de bases de citosina (de CG- a TA) y editores de bases de adenina (de AT a GC). Esta técnica no requiere RADNbc ni plantillas de ADN de donante, pero no puede determinar mutaciones de puntos de transversión30. Las transposasas Cas pueden integrar grandes secuencias genómicas en sitios diana, pero esta técnica no se ha descrito en células de mamíferos.

La edición de calidad (prime editing) es una nueva tecnología que puede establecer específicamente todos los tipos posibles de mutaciones puntuales, pequeñas inserciones y pequeñas eliminaciones. Los principales editores están compuestos por un dominio Cas9 fusionado a una transcriptasa inversa y son guiados al ADN diana por un ARN guía de edición de calidad (ARNgec) que también codifica la edición deseada. La transcriptasa inversa copia la plantilla del ARNgec en el ADN que se incorpora en el ADNbc30. Aunque es muy prometedor, este enfoque está limitado por las dimensiones del ADN modificado y por el desarrollo de estrategias de administración aceptables.
SISTEMAS DE ADMINISTRACIÓNSe emplean diversos vectores virales para administrar el material genético con eficacia.
LentivirusLos lentivirus son virus de ARN monocatenario encapsulados capaces de infectar tanto células en división como células que no se dividen31. Estos virus suministran su carga genética al integrarla en el genoma de la célula huésped y crean una respuesta duradera dentro de la célula infectada y las células descendientes posteriores31. Sin embargo, este mecanismo aumenta el potencial de oncogénesis de inserción, un evento adverso que se ha atenuado con el desarrollo de vectores lentivirales autoinactivantes32–34.
AdenovirusLos adenovirus (AV) son virus icosaédricos no encapsulados con un genoma de ADN bicatenario que puede llegar a contener grandes transgenes de 54kb35. El AV tiene un elevado índice de transducción génica, incluso en miocardiocitos, pero la expresión de proteínas puede ser breve, con una duración de 4 semanas36. El AV también puede producir una respuesta inflamatoria considerable y la presencia de anticuerpos preexistentes surgidos de la exposición previa al AV de referencia puede disminuir su eficacia en la transducción37.
Virus adenoasociadosEl VAA es un virus no encapsulado con un genoma de ADN monocatenario con la replicación alterada36. A diferencia de los VAA de referencia, los VAA recombinantes (VAAr) no se integran en los cromosomas humanos y el genoma viral continúa siendo extracromosómico en una forma episómica circular38. Algunos serotipos de VAA tienen un tropismo natural por tejidos específicos, como el VAA9 por el hígado y el corazón. Aunque los VAA son menos inmunogénicos que los AV, todavía tienen el potencial de activación inmunitaria y una mayor depuración por parte de anticuerpos neutralizantes (AcN) preexistentes36,39. La capacidad por el tamaño de los genes de los VAA se limita a 4,7kb. Por lo tanto, se está desarrollando un enfoque que utiliza vectores VAA dobles que expresan varias secuencias genómicas que se fusionan antes de la traducción40. Los VAAr se secuestran principalmente en el hígado después de la inyección sistémica y la administración de genes a tejidos específicos, como el tejido muscular, incluido el corazón, requiere dosis elevadas del virus. Por ello se han desarrollado cápsides de VAA que contienen péptidos específicos, conocidas como MyoAAV, para mejorar la transinfección en células musculares41.
Vectores no viralesLos vectores no virales se han creado para superar las limitaciones de los vectores virales y consisten en polímeros, lípidos, partículas inorgánicas y enfoques combinados. La mayoría de estos sistemas son catiónicos y pueden combinarse con ADN de carga negativa para generar un complejo vector-gen positivo general que puede unirse a las moléculas de carga negativa de la membrana celular e internalizarse42.
Las nanopartículas lipídicas (NPL) son una solución atractiva, ya que se pueden conjugar con ligandos de direccionamiento específico para llegar a tejidos concretos. Estas nanopartículas se pueden modificar para reducir la inmunogenicidad y son asequibles para producir a gran escala43–46. Gran parte de la investigación en torno a este novedoso vehículo se centra en la administración de terapias con ARN. Las NPL se han utilizado recientemente en la modificación del genoma, pero su eficacia en la transducción de miocardiocitos es limitada47,48.
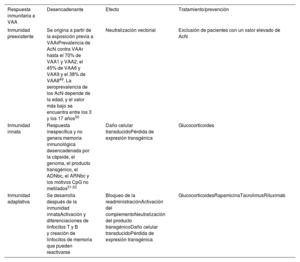
RIESGOS DE TOXICIDAD DE LOS VECTORES DE VIRUS ADENOASOCIADOSLa respuesta inmunitaria a los vectores de VAA puede causar varios eventos adversos, como hepatotoxicidad y microangiopatía trombótica. Los mecanismos inmunológicos y los posibles tratamientos inmunosupresores se presentan en la tabla 1.
Respuesta inmunitaria al virus adenoasociado
| Respuesta inmunitaria a VAA | Desencadenante | Efecto | Tratamiento/prevención |
|---|---|---|---|
| Inmunidad preexistente | Se origina a partir de la exposición previa a VAArPrevalencia de AcN contra VAAr hasta el 70% de VAA1 y VAA2, el 45% de VAA6 y VAA9 y el 38% de VAA849. La seroprevalencia de los AcN depende de la edad, y el valor más bajo se encuentra entre los 3 y los 17 años50 | Neutralización vectorial | Exclusión de pacientes con un valor elevado de AcN |
| Inmunidad innata | Respuesta inespecífica y no genera memoria inmunológica desencadenada por la cápside, el genoma, el producto transgénico, el ADNbc, el ARNbc y los motivos CpG no metilados51,52 | Daño celular transducidoPérdida de expresión transgénica | Glucocorticoides |
| Inmunidad adaptativa | Se desarrolla después de la inmunidad innataActivación y diferenciaciones de linfocitos T y B y creación de linfocitos de memoria que pueden reactivarse | Bloqueo de la readministraciónActivación del complementoNeutralización del producto transgénicoDaño celular transducidoPérdida de expresión transgénica | GlucocorticoidesRapamicinaTacrolimusRituximab |
AcN: anticuerpos neutralizantes; ADNbc: ADN bicatenario; ARNbc: ARN bicatenario; VAAr: virus adenoasociado de referencia.
La hepatotoxicidad es el evento adverso más frecuente en relación con la administración sistémica de vectores de VAA. Este efecto adverso puede presentarse como enzimas hepáticas (AST, ALT) elevadas e insuficiencia hepática. En ensayos clínicos que evaluaron la seguridad y la eficacia de los VAA portadores del transgén FIX para tratar la hemofilia B53,54, la expresión del transgén disminuyó gradualmente después de 4-8 semanas y se relacionó con un aumento de las transaminasas y la detección de células T CD8+específicas de VAA. El evento adverso fue limitado, excepto en los pacientes con AcN, y mejoró la expresión hepática del transgén, y el aumento de las transaminasas se controló con una pauta de esteroides en descenso54.
Microangiopatía trombóticaLa microangiopatía trombótica se caracteriza por lesiones en las arteriolas y los capilares, lo que conduce a una trombosis microvascular con manifestaciones clínicas como anemia hemolítica, trombocitopenia y lesión renal aguda. La microangiopatía trombótica mediada por el complemento (síndrome hemolítico urémico atípico [SHUa]) es un efecto de clase en función de la dosis de la administración intravenosa (i.v.) de VAA para tejido cardiaco y muscular. El mecanismo conlleva un rápido aumento de la inmunoglobulina M (IgM), unión a la cápside y activación de la vías clásica y alternativa del complemento. El SHUa se ha descrito en pacientes de ensayos clínicos de terapia génica para la atrofia muscular espinal, la distrofia muscular de Duchenne (DMD), la enfermedad de Danon y la enfermedad de Fabry (EF). En algunos casos se utilizó eculizumab —un anticuerpo monoclonal de IgG que previene la escisión de C5 en C5a y AC5b y evita la formación del complejo de ataque a la membrana— para tratar eficazmente la afección55,56. Todavía se desconoce si el uso profiláctico de eculizumab puede ser eficaz para prevenir este evento adverso.
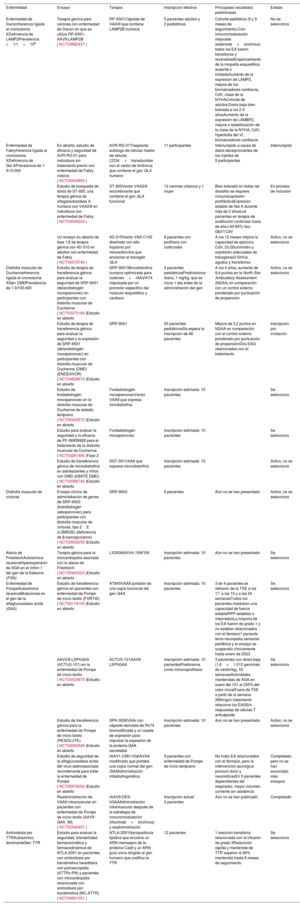
ENSAYOS CLÍNICOS QUE EVALÚAN LA TERAPIA GÉNICA EN MIOCARDIOPATÍASEstán en curso varios ensayos clínicos que evalúan la terapia génica en miocardiopatías (tabla 2). En esta sección se describen los datos preliminares de estos ensayos, con especial atención a la terapia de sustitución de genes con VAA, la técnica más utilizada en la actualidad.
Ensayos clínicos sobre terapias génicas en miocardiopatías
| Enfermedad | Ensayo | Terapia | Inscripción efectiva | Principales resultados preliminares | Estado |
|---|---|---|---|---|---|
| Enfermedad de DanonHerencia ligada al cromosoma XDeficiencia de LAMP2Prevalencia <1/1×106 | Terapia génica para varones con enfermedad de Danon en que se utiliza RP-A501; AAV9.LAMP2B (NCT03882437) | RP-A501Cápside de VAAr9 que contiene LAMP2B humana | 5 pacientes adultos y 2 pediátricos | Cohorte pediátrica (6 y 9 meses de seguimiento):Con inmunomodulación mejorada (esteroide+sirolimus) todos los EA fueron transitorios y reversiblesEmpeoramiento de la miopatía esquelética ausente o limitadoAumento de la expresión de LAMP2, mejora de los biomarcadores cardiacos, CdV, clase de la NYHACohorte de adultos:Dosis baja bien tolerada a los 2-3 añosAumento de la expresión de LAMBP2, mejora o estabilización de la clase de la NYHA, CdV, hipertrofia del VI, biomarcadores cardiacos | No se selecciona |
| Enfermedad de FabryHerencia ligada al cromosoma XDeficiencia de Gal APrevalencia de 1-5/10.000 | En abierto, estudio de eficacia y seguridad de AVR-RD-01 para individuos sin tratamiento previo con enfermedad de Fabry clásica (NCT03454893) | AVR-RD-01Trasplante autólogo de células madre de células CD34+transducidas con el vector de lentivirus que contiene el gen GLA humano | 11 participantes | Interrumpido a causa de datos decepcionantes de los injertos de 5 participantes | Interrumpido |
| Estudio de búsqueda de dosis de ST-920, una terapia génica de alfagalactosidasa A humana con VAA2/6 en individuos con enfermedad de Fabry (NCT04046224) | ST-920Vector VAA2/6 recombinante que contiene el gen GLA funcional | 12 varones clásicos y 1 mujer | Bien tolerado en todas las dosisNo se requiere inmunosupresión profilácticaExpresión estable de Gal A durante más de 2 añosLos pacientes en terapia de sustitución continúan fuera de ella↓40-65% liso-Gb3↑CdV | En proceso de inclusión | |
| Un ensayo en abierto de fase 1/2 de terapia génica con 4D-310 en adultos con enfermedad de Fabry (NCT04519749) | 4D-310Vector VAA C102 diseñado con alto tropismo por miocardiocitos que encierran el transgén GLA | 6 pacientes con profilaxis con corticoides | A los 12 meses mejora la capacidad de ejercicio, CdV, DLGSuministro y expresión adecuadas de transgenes3 SHUa agudos y transitorios | Activo, no se selecciona | |
| Distrofia muscular de DuchenneHerencia ligada al cromosoma XGen DMDPrevalencia de 1-9/100.000 | Estudio de terapia de transferencia génica para evaluar la seguridad de SRP-9001 (delandistrogén moxeparvovec) en participantes con distrofia muscular de Duchenne (NCT03375164)Estudio en abierto | SRP-9001Microdistrofina humana optimizada para codones+rAAVrh74 impulsada por un promotor específico del músculo esquelético y cardiaco | 4 pacientes pediátricosPrednisolona diaria, 1 mg/kg, que se inicia 1 día antes de la administración del gen | A los 4 años, aumento de 9,4 puntos en la North Star Ambulatory Assessment (NSAA) en comparación con un control externo ponderado por puntuación de propensión | Activo, no se selecciona |
| Estudio de terapia de transferencia génica para evaluar la seguridad y la expresión de SRP-9001 (delandistrogén moxeparvovec) en participantes con distrofia muscular de Duchenne (DMD) (ENDEAVOR) (NCT04626674)Estudio en abierto | SRP-9001 | 20 pacientes pediátricosSe espera la inscripción de 46 pacientes | Mejora de 3,2 puntos en NSAA en comparación con el control externo ponderado por puntuación de propensiónDos EAG relacionados con el tratamiento | Inscripción por invitación | |
| Estudio de fordadistrogén movaparvovec en la distrofia muscular de Duchenne de estadio temprano (NCT05429372)Estudio en abierto | Fordadistrogén movaparvovecVector VAA9 que expresa minidistrofina | Inscripción estimada: 10 pacientes | Se selecciona | ||
| Estudio para evaluar la seguridad y la eficacia de PF-06939926 para el tratamiento de la distrofia muscular de Duchenne (NCT04281485)Fase 3 | Fordadistrogén movaparvovec | Inscripción estimada: 10 pacientes | Se selecciona | ||
| Estudio de transferencia génica de microdistrofina en adolescentes y niños con DMD (IGNITE DMD) (NCT03368742)Estudio en abierto | SGT-001VAA9 que expresa microdistrofina | Inscripción estimada: 10 pacientes | Activo, no se selecciona | ||
| Distrofia muscular de cinturas | Ensayo clínico de administración de genes de SRP-9003 (bidridistrogén xeboparvovec) para participantes con distrofia muscular de cinturas, tipo 2E (LGMD2E) (deficiencia de β-sarcoglucanos)(NCT03652259)Estudio en abierto | SRP-9003 | 6 pacientes | Aún no se han presentado | Activo, no se selecciona |
| Ataxia de FriedreichAutosómica recesivaHiperexpansión de AGA en el intrón 1 del gen de la frataxina (FXN) | Terapia génica para la miocardiopatía asociada con la ataxia de Friedreich (NCT05445323)Estudio en abierto | LX2006AAVrh.10hFXN | Inscripción estimada: 10 pacientes | Aún no se han presentado | Se selecciona |
| Enfermedad de PompeAutosómica recesivaMutaciones en el gen de la alfaglucosidasa ácida (GAA) | Estudio de transferencia génica en pacientes con enfermedad de Pompe de inicio tardío (FORTIS) (NCT04174105)Estudio en abierto | AT845VAA8 portador de una copia funcional del gen GAA | Inscripción estimada: 10 pacientes | 3 de 4 pacientes se retiraron de la TSE a las 17, a las 10 y a las 24 semanasTodos los pacientes mostraron una capacidad de fuerza estableRPP estables o mejoradosLa mayoría de los EA fueron de grado 1 y no estaban relacionados con el fármaco1 paciente tenía neuropatía sensorial periférica y el ensayo se suspendió clínicamente hasta enero de 2023 | Se selecciona |
| AAV2/8-LSPhGAA (ACTUS-101) en la enfermedad de Pompe de inicio tardío (NCT03533673)Estudio en abierto | ACTUS-101AAV8-LSPhGAA | Inscripción estimada: 10 pacientesPrednisona como inmunoprofilaxis | 3 pacientes con dosis baja (1,6×1.012 genomas de vector/kg), 52 semanasActividades mantenidas de AGA en suero del 101 al 235% del valor inicialFuera de TSE a partir de la semana 26Ningún tratamiento relaciona los EAGSin respuestas de células T anticápside | Se selecciona | |
| Estudio de transferencia génica para la enfermedad de Pompe de inicio tardío (RESOLUTE) (NCT04093349)Estudio en abierto | SPK-3006VAAr con cápside derivada de Rh74 biomodificada y un casete de expresión para impulsar la expresión de la proteína GAA secretable | Inscripción estimada: 10 pacientes | Aún no se han presentado | Activo, no se selecciona | |
| Estudio de seguridad de la alfaglucosidasa ácida del virus adenoasociado recombinante para tratar la enfermedad de Pompe (NCT00976352)Estudio en abierto | rAAV1-CMV-hGAAVAA modificado que portaba una copia normal del gen GAAAdministración intradiafragmática | 9 pacientes con enfermedad de Pompe de inicio temprano | No hubo EA relacionados con el fármaco, pero la intervención quirúrgica provocó dolor y neumotóraxEn 5 pacientes dependientes del respirador, mayor volumen corriente sin asistencia | Completado, pero no se han anunciado más ensayos | |
| Readministración de VAA9 intramuscular en pacientes con enfermedad de Pompe de inicio tardío (AAV9-GAA_IM) (NCT02240407) | rAAV9-DES-hGAAAdministración intramuscular después de la estrategia de inmunomodulación (rituximab+sirolimus) y readministración | Inscripción actual: 2 pacientes | Aún no se han publicado | Completado | |
| Amiloidosis por TTRAutosómico dominanteGen TTR | Estudio para evaluar la seguridad, tolerabilidad, farmacocinética y farmacodinámica de NTLA-2001 en pacientes con amiloidosis por transtirretina hereditaria con polineuropatía (ATTRv-PN) y pacientes con miocardiopatía relacionada con amiloidosis por transtirretina (MC-ATTR) (NCT04601051) | NTLA-2001Nanopartícula lipídica que encierra un ARN mensajero de la proteína Cas9 y un ARN guía único dirigido al gen humano que codifica la TTR | 12 pacientes | 1 reacción transitoria relacionada con la infusión de grado 3Reducción rápida y mantenida de TTR superior al 90% mantenida hasta 6 meses de seguimiento | Se selecciona |
AGA: gen de la alfaglucosidasa ácida; CdV: calidad de vida; DLG: deformación longitudinal general; DMD: distrofia muscular de Duchenne; EA: eventos adversos; EAG: eventos adversos graves; Gal A: alfagalactosidasa A; GLA: gen de la alfagalactosidasa; LAMP: proteína de membrana asociada a lisosomas; LAMP2B: isoforma B de proteína de membrana asociada a lisosomas 2; Liso-Gb3: globotriaosilesfingosina; NSAA: North Star Ambulatory Assessment; NYHA: New York Heart Association; RPP: resultados percibidos por el paciente; SHUa: síndrome hemolítico urémico atípico; TSE: terapia de sustitución enzimática; TTR: transtirretina; VAA: virus adenoasociado; VAAr: virus adenoasociado recombinante; VI: ventrículo izquierdo.
La enfermedad de Danon es un trastorno lisosómico raro ligado al cromosoma X y causado por variantes patogénicas del gen LAMP2 (proteína de membrana asociada a lisosomas 2) que origina una alteración de la autofagia y la acumulación de vacuolas autofágicas en el tejido muscular. Entre las manifestaciones clínicas se incluye una miocardiopatía hipertrófica (MCH) rápidamente progresiva, con una supervivencia media de 19 años para los varones en ausencia de trasplante cardiaco57. No hay ningún tratamiento farmacológico capaz de modificar el curso de esta enfermedad. Se ha evaluado en un ensayo abierto fase 158 una infusión i.v. única de RP-A501, un vector VAA9 que expresa LAMP2B, a una dosis baja (6,7×1013 GC/kg) o a una dosis alta (1,1×1014 GC/kg) en varones de 8 a 14 años y de 15 años o mayores. Los resultados preliminares del estudio se presentaron en la reunión científica anual de 2022 de la Heart Failure Society of America. En 3 pacientes de la cohorte 1 (edad ≥ 15 años, dosis baja), el RP-A501 aumentó la expresión de LAMP2B medida mediante inmunoelectrotransferencia e inmunoquímica en el tejido miocárdico. Los 2 pacientes con cumplimiento constatado del régimen inmunosupresor mostraron una alta expresión cardiaca de LAMP2B (el 67,8 y el 92,4% frente al control normal por inmunoquímica) y mejoras de la clase funcional de la New York Heart Association (NYHA) y de péptidos natriuréticos. Los 3 pacientes mostraron mejora o estabilidad de la capacidad funcional medida por la prueba de 6 min de marcha, un aumento de la actividad física y una disminución de las vacuolas autofágicas en la biopsia endomiocárdica. Entre los eventos adversos más frecuentes se encontraban disminución plaquetaria reversible, aumento de transaminasas y miopatía esteroidea. En la cohorte de adultos con dosis alta, 1 paciente presentó microangiopatía trombótica mediada por el complemento con trombocitopenia y lesión renal aguda que requirió hemodiálisis temporal. Por consiguiente, no se ha incluido a más pacientes en la cohorte de dosis alta; además, se ajustó el tratamiento inmunosupresor limitando la dosis diaria de corticoides, utilizando sirolimus en lugar de tacrolimus para minimizar el impacto renal y continuando con rituximab. En septiembre de 2022 se presentó una actualización de los resultados del ensayo de fase 159. En la cohorte de adultos, se confirmó una reducción considerable de los biomarcadores, la reducción del grosor de la pared del ventrículo izquierdo, la mejora o estabilidad de la clase de la NYHA y de la puntuación del Kansas City Cardiomyopathy Questionnaire hasta los 36 meses de seguimiento. En los 2 pacientes de la cohorte pediátrica, con una mejora del protocolo de inmunomodulación, no hubo constancia de trombocitopenia, miopatía esquelética, elevación de transaminasas tardía o eventos adversos relacionados con la activación del complemento. La expresión de proteínas mejoró en el último seguimiento a los 3 y a los 6 meses. Además, se observó una mejora en los biomarcadores, los síntomas comunicados y la clase de la NYHA a los 6 y a los 9 meses. La expresión de LAMP2 aumentó de manera similar que en la cohorte de adultos en el mismo periodo de tiempo y el área vacuolar en la biopsia endomiocárdica disponible se redujo de manera considerable. Sobre la base de estos resultados positivos, la FDA estadounidense ha otorgado al RP-A501 la designación de terapia avanzada de medicina regenerativa, y está previsto que la fase 2 del ensayo comience el segundo trimestre de 202360.
Enfermedad de FabryLa EF es un trastorno de almacenamiento lisosómico ligado al cromosoma X causado por variantes patógenas del gen GLA (alfagalactosidasa). Esta alteración produce diversos grados de carencia de alfagalactosidasa (Gal A) y acumulación de glucoesfingolípidos, predominantemente globotriaosilceramida (Gb3) y su derivado desacilado globotriaosilesfingosina (liso-Gb3), en órganos diana como el corazón, los riñones y los vasos sanguíneos. El procedimiento diagnóstico habitual comprende el tratamiento de sustitución enzimática i.v. o el tratamiento oral con una chaperona que, aunque es eficaz para mejorar la calidad de vida, el dolor neuropático, las manifestaciones gastrointestinales y la función renal, tiene una eficacia limitada en la miocardiopatía61.
La EF es un candidato adecuado para la terapia génica, ya que es una enfermedad monogénica y se pueden lograr efectos clínicos con un 10% de actividad de cantidades normales de Gal A. Varios estudios están evaluando actualmente la seguridad y la eficacia de la terapia génica en la EF.
AVR-RD-01En un estudio abierto que evaluó el AVR-RD-01, se utilizó el trasplante autólogo de células madre de células CD34+transducidas con un vector de lentivirus que contenía el gen humano GLA. Sin embargo, se ha detenido la inscripción en el ensayo debido a los decepcionantes datos de injerto en 5 participantes62.
ST-920Por medio del ST-920 (isaralgagén civaparvovec), el gen GLA funcional se administra al hígado a través de una infusión única de un vector VAA2/6 recombinante sin necesidad de tratamiento inmunosupresor. Este enfoque se encuentra actualmente en estudio en el ensayo STAAR de fase 1/2 (NCT04046224). En la fase de aumento de la dosis del ensayo, se incluyó a 9 varones con EF clásica de gravedad leve/moderada. En la fase de ampliación, 5 pacientes recibieron una dosis de 5×1013 vg/kg. A partir de octubre de 2022, fecha límite para la inclusión de datos, no se han notificado eventos adversos por el tratamiento superiores al grado 2 en los 13 pacientes del estudio.
Los 9 pacientes tratados en la fase de aumento de la dosis en las 4 cohortes de dosis (0,5×1013, 1×1013, 3×1013 y 5×1013 vg/kg) mostraron actividad de Gal A según la dosis, suprafisiológica y mantenida que varió de casi 4 veces a 68 veces la concentración normal media del paciente con el seguimiento más prolongado durante más de 2 años. Se observó una reducción estable y según la dosis en la concentración de Lysogb3 a valores basales altos (> 80 ng/ml) y, a la dosis más alta, también a una concentración plasmática baja (< 25 ng/ml) hasta los 25 meses de seguimiento. El tratamiento también se relacionó con una mejora potencial de la nefropatía de Fabry. De hecho, se ha notificado una reducción de las inclusiones de Gb3 y la pérdida de podocitos en el paciente 9.
C102Se descubrió que el C102, un vector de VAA modificado con un elevado tropismo por miocardiocitos, transduce el tejido cardiaco de primates no humanos in vivo junto con miocardiocitos humanos y células endoteliales in vitro mejor que los vectores serotipados con VAA1, VAA8 o VAA9, lo que permite la administración intravenosa de dosis bajas63.
El 4D-310 (vector C102 que encierra el transgén GLA) promueve concentraciones sanguíneas de Gal A altas y estables que las células cercanas pueden captar, pero también permite la transducción directamente a las células del tejido diana, como los miocardiocitos, los riñones (glomérulos incluyendo los podocitos) y los vasos sanguíneos64.
Los ensayos clínicos en abierto de fase 1/2 INGLAXA-1 y -2 evalúan actualmente la seguridad y la tolerabilidad del 4D-310 en pacientes con EF clásica o de inicio tardío con afección cardiaca dentro o fuera del tratamiento de sustitución enzimática con inmunosupresión profiláctica con corticoides. En la cohorte 1 (113 vg/kg con tratamiento inmunológico con corticoides) se incluyó a 6 pacientes; 4 tenían el antecedente de tratamiento de sustitución enzimática y 1 seguía tratamiento con chaperona. La deformación longitudinal global y el VO2 máximo mejoraron desde el inicio hasta la semana 52 en 2 de 3 pacientes. A 1 paciente se le realizó una biopsia cardiaca la semana 6 y el tejido se demostró sano, sin signos de inflamación. La hibridación in situ mostró captación solo en miocardiocitos, con alrededor del 50% de los miocardiocitos positivos en la expresión transgénica. En inmunohistoquímica, la proteína AGA se expresó en todas las muestras. La administración al genoma fue satisfactoria, con 4,4 copias de vector por miocardiocito y la expresión del ARN fue de 16,2 copias de transcrito por miocardiocito.
Tres pacientes sufrieron un SHUa agudo y transitorio, con inicio entre los días 3 y 7; 1 paciente no requirió intervención, otro fue tratado con eculizumab y otro más fue tratado con eculizumab y diálisis transitoria. Después del efecto limitante de dosis por toxicidad del SHUa, la FDA paralizó el programa clínico.
Se procedió a evaluar a fondo la respuesta inmunitaria de los pacientes y se encontró que aquellos que presentaron SHUa mostraron activación del complemento antes de la dosificación. Los próximos pasos de este ensayo comprenden la armonización con la FDA para resolver la suspensión clínica y reanudar la inclusión después de modificar el protocolo, con la implementación de un tratamiento inmunosupresor con rituximab/sirolimus, la adición de una preselección según el complemento y la exclusión de pacientes con activación del complemento antes de la dosificación.
Distrofia muscular de DuchenneEl gran tamaño del gen DMD plantea retos a la posible aplicación de terapias de transferencia génica. Sin embargo, una estructura del gen más pequeña podría paliar el fenotipo de la enfermedad y se han creado varios genes recombinantes que codifican mini/microdistrofina65.
Un ensayo clínico de fase 3 sobre el fordadistrogén movaparvovec (SRP-9001), un vector de VAA9 que expresa minidistrofina, está en curso después de una suspensión clínica inicial de la FDA a causa de la muerte de un participante (NCT04281485)66.
El SRP-9001 está formado por VAAr rh74 (rAAVrh74) y una microdistrofina humana optimizada por codones impulsada por un promotor específico del músculo esquelético y cardiaco, que mejora la expresión cardiaca67. Los resultados preliminares del ensayo controlado no aleatorizado de fase 1/2a han mostrado una mejora de las capacidades motrices funcionales en 4 pacientes pediátricos después de 4 años y está en curso un ensayo de fase 368.
La técnica de omisión de exones (exon skipping) permite el silenciamiento de exones específicos que determinan la restauración del marco de lectura de un gen mutado mediante el silenciamiento de ARN, oligonucleótidos antisentido o modificación del genoma. La omisión de exones es una técnica específica para determinadas mutaciones y en DMD conduce a la producción de proteína distrofina acortada, pero funcional. La FDA ha aprobado 4 terapias de oligonucleótidos antisentido para el tratamiento de DMD y se está estudiando un vector de VAA que administra ARN nuclear pequeño que contiene secuencias antisentido dirigidas al exón 2 de DMD duplicado65.
Ataxia de FriedreichUn estudio de fase 1A/1B evalúa actualmente la seguridad y la eficacia de un serotipo rh.10 VAA que administra un gen de frataxina funcional (FXN) para el tratamiento de la miocardiopatía por ataxia de Friedreich (NCT05445323). En un modelo murino, este enfoque redujo la masa cardiaca, mejoró la función cardiaca, corrigió los defectos bioquímicos relacionados con FXN y redujo la disfunción cardiaca inducida por el estrés69,70.
El complejo ribonucleoproteico para la escisión de las secuencias repetidas de alfaglucosidasa ácida (AGA) en FXN ha mostrado resultados prometedores en un modelo murino y un ensayo clínico se encuentra en la fase de planificación71.
Enfermedad de PompeVarias terapias génicas diseñadas para facilitar una copia funcional del gen GAA se encuentran en diversas etapas de desarrollo.
En 2015, se realizó el primer ensayo con seres humanos de terapia génica diafragmática (VAA1-CMV-AGA) para tratar la disfunción respiratoria y neural de la enfermedad de Pompe de inicio temprano. No se han notificado eventos adversos relacionados con el fármaco, pero la inyección intradiafragmática provocó neumotórax y contusión pulmonar, y el rendimiento ventilatorio solo mejoró de manera discreta.
El análisis inicial de 3 pacientes con enfermedad de Pompe de inicio tardío en el ensayo de ACTUS-101 y el vector VAA8 que expresa AGA humana con el control de transcripción de un promotor específico del hígado (VAA2/8-LSPhAGA) mostró un aumento mantenido de la AGA y ningún evento adverso grave relacionado con el tratamiento72.
El AT845 es un VAA8 diseñado para expresar AGA humana específicamente en el músculo esquelético y el corazón, y actualmente está en estudio en pacientes con enfermedad de Pompe de inicio tardío, con resultados preliminares prometedores73.
AMILOIDOSIS POR TRANSTIRRETINASe está evaluando el sistema CRISPR-Cas9 para el tratamiento de la amiloidosis por transtirretina (TTR). La amiloidosis por TTR se caracteriza por el depósito progresivo de TTR nativa o mutada mal plegada en varios tejidos, como el corazón y el sistema nervioso periférico. NTLA-2001 se incluye en una NPL opsonizada por apolipoproteína E que actúa como sistema portador. Los componentes activos son un ARN mensajero de una proteína Cas9 y un ARN guía (ARNg) único dirigido al gen humano que codifica la TTR. Las NPL de NTLA-2001 se administran por vía i.v. y llegan al hígado, donde son absorbidas por el receptor de lipoproteínas de baja densidad expresado por los hepatocitos. La molécula de ARN de Cas9 se traduce en el citoplasma y produce la enzima endonucleasa Cas9. Junto con la endonucleasa Cas9, el ARNg único específico de TTR forma un complejo ribonucleoproteico CRISPR-Cas9. Este último entra en el núcleo, el ARNg único se une al ADN diana y el complejo CRISPR-Cas9 puede desenrollar la hélice y alcanzar el gen diana. El gen TTR finalmente se escinde y la corrección del ADN endógeno a través de la unión de extremos no homólogos introduce inserciones o eliminaciones de bases («indels») que provocan la reducción de ARNm funcional y TTR. En hepatocitos humanos, el NTLA-2001 produjo saturación de la edición de TTR (≥ 93,7%), lo que conllevó una reducción de al menos el 91% de la expresión de ARNm de TTR y proteína TTR. La secuenciación de próxima generación mostró que el NTLA-2001 indujo la inactivación de TTR. Estudios preclínicos en ratones transgénicos y macacos cangrejeros mostraron que la edición de TTR causó una reducción mantenida de la TTR durante un periodo de 12 meses. Un estudio de viabilidad en abierto, de dosis única, ha incluido a 6 pacientes, de 46 a 64 años, con polineuropatía por amiloidosis por TTR hereditaria. A todos los pacientes se les administró la infusión y en 1 paciente apareció una reacción de grado 1 relacionada con la infusión. Los valores de coagulación y las cifras de plaquetas y transaminasas se mantuvieron dentro de los intervalos de referencia. Se observó una reducción rápida y mantenida en función de la dosis de la concentración de TTR, con una reducción el día 28 entre el 47 y el 56% en el grupo de dosis más baja (0,1mg/kg) y entre el 80 y el 96% en el grupo de dosis más alta (0,3mg/kg)47.
Se han presentado datos en 12 pacientes con miocardiopatía por amiloidosis por TTR hereditaria o nativa en la fase de aumento de dosis (9 pacientes con 0,7mg/kg, 3 pacientes con 1,0mg/kg). El NTLA-2001 fue bien tolerado y hubo una sola reacción de grado 3 relacionada con la infusión, que se resolvió sin secuelas clínicas. Todos los pacientes lograron una reducción de la concentración de TTR superior al 90% a los 28 días, que se mantenía a los 6 meses de seguimiento74.
OBJETIVOS FUTUROSMYH7También se están estudiando modelos preclínicos para la modificación del genoma mediante CRISPR-Cas9 en casos de miocardiopatía hipertrófica (MCH). La MCH se define por un grosor del ventrículo izquierdo superior a 15mm y la enfermedad puede estar causada por variantes patogénicas en genes que codifican proteínas sarcoméricas. Entre las principales manifestaciones clínicas se encuentran la reducción de la capacidad de ejercicio, insuficiencia cardiaca y arritmias. Se han propuesto varios enfoques. Chai et al.75 utilizaron un sistema de administración doble de VAA9 que incluía una edición de base de adenina (ABEmax-VRQR, que interviene en la conversión de A•T en G•C sin escisión del ADN) y un ARN guía (h403_sgRNA) para dirigirse a la variante patogénica de sentido alterado p.Arg403Gln en el gen MYH7. El tratamiento corrigió la adenina patogénica diana con una eficacia del 98% en células madre pluripotentes inducidas que provienen de pacientes con MCH con una edición mínima por vecindad de adeninas cercanas y una baja edición de ADN colateral. Los miocardiocitos diferenciados tenían una fuerza contráctil y una energía celular normales en comparación con los miocardiocitos no corregidos. En un modelo de ratón humanizado de MCH, la inyección intratorácica de una dosis única de VAA9 doble que codifica ABEmax-VRQR y h403_sgRNA produjo la corrección del alelo patogénico, una reducción de los transcritos patogénicos y una reducción de la hipertrofia cardiaca y el remodelado histopatológico.
Reichart et al.40 utilizaron una estrategia doble similar con VAA para administrar una ABE y ARNg para corregir la misma variante patogénica en un modelo de ratones no humanizados y obtuvieron una corrección de la variante en más del 70% de los miocardiocitos ventriculares, con lo que se evitó la aparición del fenotipo patológico. Como enfoque alternativo, se probó la administración de VAA9 de nucleasa Cas9 guiada por ARN. La técnica inactivó eficazmente el alelo patogénico, pero con dosis mayores también se redujo la función contráctil, lo que demuestra la edición accidental del alelo nativo.
MYBPC3Las mutaciones de pérdida de función en la proteína de unión a miosina C3 (MYBPC3) junto con MYH7 son las causas genéticas de MCH más frecuentes. La reducción de MYBPC3 en el sarcómero finalmente produce un aumento de las cabezas de miosina disponibles para la interacción con actina e hipercontractilidad.
Una sola administración de VAA9-MYBPC3 en ratones homocigotos con inserción génica dirigida a MYBPC3 que imitan una forma neonatal de miocardiopatía hipertrófica impidió la aparición de hipertrofia cardiaca durante 34 semanas de observación y, en función de la dosis, aumentó el ARNm de MYBPC3 y la proteína. Además, el tratamiento redujo inesperadamente la concentración de especies de ARNm patológicas. Probablemente, dado que la proteína MYBPC transgénica se integraría de manera más estable en el sarcómero, un mecanismo de retroalimentación puede reducir la transcripción de la proteína mutante76. En otro estudio, dado que el dominio aminoterminal de MYBPC3 está implicado en las interacciones con actina y miosina, la transferencia del único dominio aminoterminal de MYBPC a través de VAA9 en comparación con la secuencia completa se ha evaluado en un modelo murino sin cMyBPC (cMyBPC–/–). La transferencia del dominio aminoterminal fue suficiente para prevenir el fenotipo de la MCH macroscópico (hipertrofia ventricular izquierda) y microscópico (trastorno miocárdico y fibrosis). Con todo, aún se desconoce si la terapia con VAA puede rescatar el fenotipo de la MCH e inducir remodelado inverso en la MCH manifiesta77. El inicio de un ensayo clínico de fase 1 para evaluar la seguridad, la tolerabilidad y la eficacia clínica de una sola infusión i.v. de una terapia génica de VAA para el tratamiento de la MCH relacionada con la mutación de MYBPC3 se ha programado para el tercer trimestre de 202378.
BAG3El gen BAG3 (atanogén 3 asociado con BCL2) está muy expresado en el corazón, el músculo esquelético y el sistema nervioso central. El BAG3 en miocardiocitos mejora la contractilidad al vincular el receptor betaadrenérgico y el canal de Ca2+de tipo L, facilita la autofagia al interactuar con las proteínas de choque térmico, ofrece soporte al sarcómero al vincular la actina con el disco Z y tiene propiedades antiapoptóticas a través de la unión a BCL279. Las variantes de truncamiento o eliminación en el gen BAG3 que reducen la proteína BAG3 se han asociado con miocardiopatía dilatada (MCD)80. Los miocardiocitos humanos de MCD muestran función contráctil del sarcómero disminuida y recambio alterado de miofilamentos con proteínas mal plegadas/disfuncionales que permanecen integradas en el sarcómero. Además, la expresión de miofilamentos de BAG3 disminuye en la MCD y se correlaciona con la reducción de la función contráctil. En ratones que sufrieron un infarto de miocardio, la administración del vector VAA9 recombinante que expresa el gen BAG3 de ratón restableció la función cardiaca y el recambio de miofilamentos81. Además, la administración i.v. del VAA9-BAG3 a ratones haploinsuficientes de Bag3 evitó el inicio de la fracción de eyección reducida82.
En un informe reciente, la infusión del seno coronario mediante catéter por acceso retrógrado se utilizó de modo seguro para administrar dosis bajas de VAA9-BAG3 en minicerdos sanos, lo que produjo una transducción difusa al miocardio83. Un ensayo clínico de fase 1/2 para evaluar la seguridad y la eficacia de una sola administración mediante infusión del seno coronario por acceso retrógrado en pacientes con fracción de eyección reducida y haploinsuficiencia de BAG3 está en la fase de planificación60.
PKP2La miocardiopatía arritmogénica es una enfermedad miocárdica que se caracteriza por la sustitución fibrograsa del ventrículo derecho, el izquierdo o ambos. Las modificaciones estructurales crean un medio proarrítmico y determinan la dilatación progresiva de la cavidad y la disfunción sistólica. La miocardiopatía arritmogénica en el 40-50% de los casos se asocia con mutaciones en los genes desmosómicos, y la placofilina 2 (PKP2) es la más frecuente. Se ha evaluado un vector VAArh10 que expresa el gen PKP2 (LX2020) en ratones mutantes de PKP2. LX2020 mostró una mejora según de la dosis en la supervivencia de los ratones, una reducción de los latidos ectópicos y una mejora de la fracción de eyección izquierda y derecha84. También se han comunicado datos preclínicos sobre ratones con PKP2 inactivado para TN-401 (vector VAA9 que expresa PKP2), que muestran una reducción de las arritmias ventriculares, prevención de la disfunción ventricular derecha e izquierda y prolongación de la esperanza de vida. Además, la terapia demostró ser eficaz para restablecer los desmosomas y las uniones comunicantes y evitar la fibrosis85. Los ensayos clínicos que evalúan estas estrategias se encuentran en la fase de diseño.
PRKAG2Las mutaciones de PRKAG2 causan una miocardiopatía por acumulación de glucógeno que se caracteriza por un fenotipo hipertrófico, arritmias supraventriculares y bloqueos auriculoventriculares. Se ha utilizado un sistema CRISPR-Cas9 combinado con VAA9 para alterar el alelo de PRKAG2 mutante que codifica la mutación H503 en ratones con inserción génica. Una sola inyección sistémica del producto el día 4 o el día 42 restableció la morfología y la función en el modelo murino y redujo el grosor del ventrículo izquierdo y el contenido de glucógeno del miocardio86.
LMNAEl síndrome de progeria de Hutchinson-Gilford se caracteriza por el envejecimiento prematuro y la muerte por causas cardiovasculares, y lo causan mutaciones en el gen LMNA que se traduce en progerina, que ejerce un efecto dominante negativo. La tecnología CRISPR-Cas9 se ha utilizado para crear ratones que expresan de manera ubicua progerina y lamina C y carecen de lamina A y permite la supresión de la progerina y el restablecimiento de la lamina A de una manera específica en función del tiempo y del tipo de célula en la activación de la recombinasa Cre. La supresión de progerina y la restauración de la lamina A restringidas a las células lisas vasculares y los miocardiocitos evitaron el daño vascular y normalizaron la vida de este modelo murino87.
RETOS ACTUALESLa terapia génica podría ser un verdadero punto de inflexión en el tratamiento de distintas enfermedades; sin embargo, es necesario resolver algunos problemas. Los mayores retos con la terapia génica con VAA consisten en mejorar la eficacia de la transducción, hallar la mejor estrategia para dirigirse a tejidos específicos, reducir las dosis administradas para minimizar los posibles eventos adversos, identificar las estrategias inmunosupresoras más eficaces para amortiguar la respuesta inmunitaria y, por último, permitir una readministración del fármaco.
Los sistemas de modificación del genoma pueden tener actividad colateral, es decir, alterar el genoma en regiones distintas del sitio objetivo. Las variantes Cas de alta fidelidad pueden reducir las modificaciones colaterales, pero también se asocian con una menor actividad terapéutica30.
Aunque se han presentado varias opciones, un sistema de administración aceptable, no inmunogénico y específico para el tejido continúa siendo una necesidad sin cubrir.
La selección de pacientes sigue sin resolverse; de hecho, aún se desconoce si el tratamiento podría rescatar un fenotipo ya manifiesto o si debería administrarse en la fase temprana de la enfermedad.
Aún se desconocen la durabilidad y los efectos a largo plazo.
Otros problemas son la asequibilidad, la regulación y el acceso al tratamiento. Todos estos enfoques tienen grandes costes de fabricación y el acceso al tratamiento puede estar limitado.
CONCLUSIONESLa terapia génica es un campo prometedor y en evolución que ya permite el tratamiento dirigido de enfermedades graves, como la amaurosis congénita de Leber y la atrofia muscular espinal. Los problemas de seguridad y durabilidad aún no se han resuelto; sin embargo, se espera que varios ensayos clínicos en curso ofrezcan respuestas y amplíen la aplicación de la técnica a otras afecciones.
FINANCIACIÓNLos autores no han recibido ninguna financiación específica para este trabajo.
CONTRIBUCIÓN DE LOS AUTORESE. Adler contribuyó al trabajo con las actividades de concepción, supervisión, revisión y edición. A. Argirò y J. Ding escribieron el borrador original y realizaron modificaciones bajo la supervisión de E. Adler.
CONFLICTO DE INTERESESE. Adler participa en el Consejo Asesor Científico de Medtronic, Fuji y SanaChief. E. Adler trabaja como científico para Lexeo Therapeutics y como asesor para Abiomed, AstraZeneca, Ionis, Medtronic, Abbott y Novartis. E. Adler forma parte del consejo asesor y es accionista de Rocket Pharmaceuticals. E. Adler es fundador del consejo científico y accionista de ResQue Therapeutics. A. Argirò y J. Ding no tienen conflictos de intereses que declarar.