
Gene therapy strategies encompass a range of approaches, including gene replacement and gene editing. Gene replacement involves providing a functional copy of a modified gene, while gene editing allows for the correction of existing genetic mutations. Gene therapy has already received approval for treating genetic disorders like Leber's congenital amaurosis and spinal muscular atrophy. Currently, research is being conducted to explore its potential use in cardiology. This review aims to summarize the mechanisms behind different gene therapy strategies, the available delivery systems, the primary risks associated with gene therapy, ongoing clinical trials, and future targets, with a particular emphasis on cardiomyopathies.
Keywords
Human gene therapy exerts its impact through the transcription and/or translation of transferred genetic material, and/or by integrating into the host genome. These therapies are administered in the form of nucleic acids, viruses, or genetically engineered microorganisms. The objective of this review is to examine the present status of gene therapies, their application in the treatment of cardiovascular diseases, and future challenges, with a specific emphasis on cardiomyopathies.
HISTORY OF GENE THERAPIESHuman gene therapy has undergone rapid development since Theodore Friedmann and Richard Roblin published the first accounts on its potential and challenges in 1972.1 Since then, scientists have strived to develop a safe, long-lasting, and effective method for modifying and manipulating human cells to achieve therapeutic effects for various heritable diseases. In 1989, the first approved human gene therapy trial took place, using a retroviral vector to insert a gene encoding neomycin resistance into human tumor-infiltrating lymphocytes for the treatment of metastatic melanoma.2 Just under a year later, another group of researchers conducted the first approved gene therapy trial for heritable genetic diseases. In that study, a retroviral vector delivered a functional copy of the adenosine deaminase gene to explanted T lymphocytes obtained from patients with adenosine deaminase-deficient severe combined immune deficiency. Over a 2-year period, the 2 patients received multiple autologous infusions of corrected T cells.3 These clinical trials laid the groundwork for the potential of gene therapy by demonstrating the feasibility, durability, and potential safety of using viral vectors to deliver genetic material as a therapeutic approach.
Despite these landmark studies, the death of18-year-old Jesse Gelsinger in 1999 highlighted the dangers of gene therapy at the time. Mr. Gelsinger had partial ornithine transcarbamylase deficiency and participated in a trial aiming to use adenoviral vectors to deliver functional ornithine transcarbamylase genes. More than 3.8 x 1013 adenoviral particles were injected into his liver, triggering a cytokine storm and acute respiratory distress leading to his death.4
Clinical trials conducted throughout the 1990s and early 2000s encountered various schallenges primarily related to unforeseen long-term toxicities and immune responses. A subset of patients enrolled in these trials developed leukemic malignancies as the retroviral vector used had the potential to activate proto-oncogenes.5,6 Other patients mounted large immune responses against the viral vectors as well as to the newly transduced cells expressing the protein products of the gene therapy.7
In 2012, a bacterial defense mechanism called CRISPR/Cas9 that could be used to easily manipulate genomes, was discovered by the Nobel laureates Jennifer Doudna and Emmanuelle Charpentier laying the foundations for gene editing.8,9 A month later, the first gene therapy was approved by the European Medicines Agency (an adeno-associated virus [AAV] vector encoding lipoprotein lipase, for the treatment of lipoprotein lipase deficiency).10 In 2017, the first gene therapy was approved in the United States by the Food and Drug Administration (FDA) for the treatment of Leber congenital amaurosis.11
In 2020, the FDA released 6 guidelines, standardizing and streamlining the drug approval process for new gene therapies.12 In response to the SARS-CoV-2 pandemic, gene therapy achieved a major breakthrough with the approval and adoption of 2 messenger RNA (mRNA)-based vaccines. These vaccines used novel lipoprotein nanoparticles to safely deliver the genetic payload into the cell cytoplasm.13,14 Currently, 27 approved gene therapies are available in the United States with dozens of clinical trials underway.15
In the field of cardiology, some pioneering studies of gene therapy were first performed in 1990, in which a murine retrovirus was used to deliver a recombinant beta-galactosidase gene into the wall of an arterial segment.16 In 1996, Dr Jeffrey Isner used a plasmid containing vascular endothelial growth factors, which was suspended in a hydrogel polymer and coated onto an angioplasty balloon that was applied to the distal popliteal artery to induce angiogenesis.17 This approach laid the foundation for a series of clinical trials aimed at injecting naked plasmid DNA encoding for angiogenic factors like vascular endothelial growth factor into or at the walls of coronary and peripheral arteries, as well as into the myocardium itself.18–20 However, none of these trials had a significant impact on clinical outcomes.
In 2007, the first gene therapy clinical trial for patients with heart failure aimed to determine the effects of delivering a gene expressing SERCA2a through an AAV to improve calcium handling within cardiomyocytes where the Ca2+ ATP-ase was downregulated.21 Unfortunately, the treatment failed to improve clinical outcomes.21,22
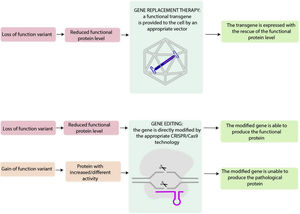
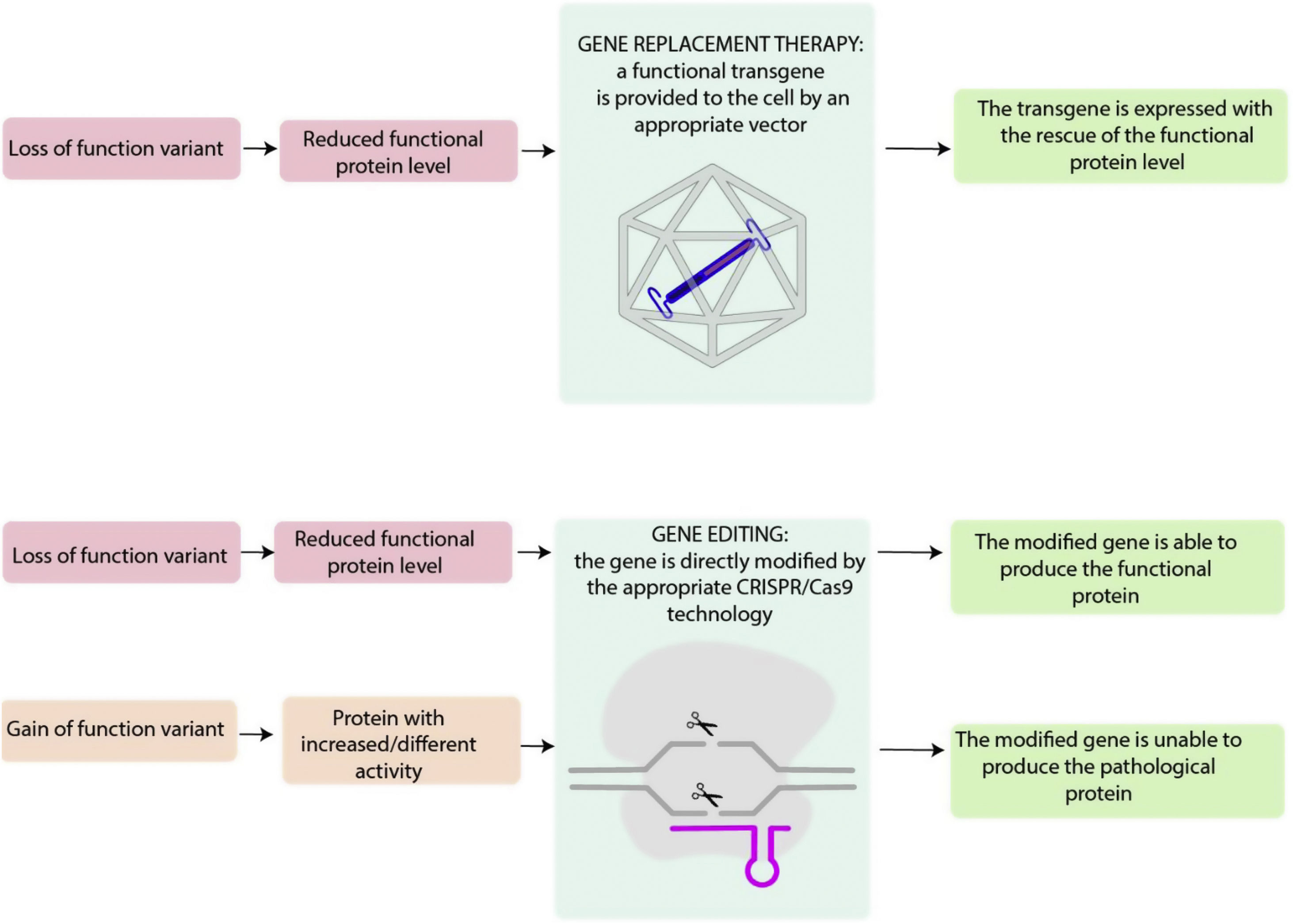
TYPES OF GENE THERAPIESIn genetic diseases, loss-of-function mutations, usually nonsense, splice, frameshift or missense, may lead to a lack of protein production or to the release of a nonfunctional protein. Gain-of-function mutations, typically from missense variants, determine the production of a protein with increased or different activity compared with the wild-type form.
Appropriate gene therapy approaches may target these alterations (figure 1). It is helpful to break down the different gene therapies into 2 categories based on their general treatment goals. Gene silencing through RNA interference or antisense oligonucleotide are not strictly considered to be gene therapy and are therefore not discussed in this manuscript.

Central illustration. Loss-of-function variants result in a diminished level of functional proteins, which can potentially be addressed through gene replacement therapy. This approach involves introducing a functional transgene into the affected cell, allowing for the restoration of the proper expression of functional protein. Genome editing includes several techniques that allow direct modification of the endogenous genome and, in the case of loss-of-function variants, may restore the functional protein, and, in the case of gain-of-function variant, may impede pathological protein expression.
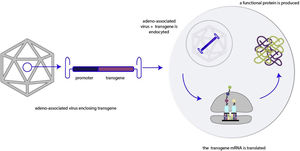
Gene replacement aims to replace a defective gene with a functional copy to achieve a therapeutic effect mitigating the effects of a loss-of-function mutation (figure 2).23 Gene replacement products can include plasmid DNA or genetic material enclosed in viral vectors, which allow the transduction of a foreign copy of the gene of interest into the target cell.24,25 Plasmid-based gene therapy may potentially produce a durable effect while minimizing the genetic disruption of the cell's genome, resulting in a lower oncogenic potential compared with viral vector-based gene therapies.26 However, plasmid delivery to the nucleus is difficult and protein expression may not be durable, especially in dividing cells.27,28 Viral vector-based gene therapy is able to produce durable expression of gene products,29 but this approach has toxicity risks that will be evaluated later in this article.

An appropriate vector, in this case an adeno-associated virus, carries the transgene. The complex AAV+transgene is endocyted by the cell and the transgene is transcripted and translated, allowing the production of a functional protein. AAV, adeno-associated virus; mRNA, messenger RNA.
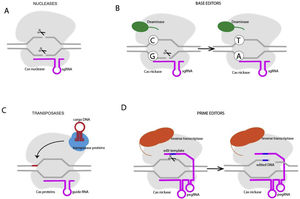
Unlike gene replacement therapy, in which a new gene is provided through an appropriate vector, genome editing consists of modifying the cell's endogenous genomic sequence and could be used for both loss and gain-of-function mutations. The type of edits desired may be obtained through CRISPR/Cas9 technology and include: conversion of DNA base pairs, deletion of DNA base pairs, insertion of DNA base pairs or a combination of the abvoe (figure 3). CRISPR/Cas9 includes 2 fundamental components: a guide RNA to match a target gene (CRISPR, clustered regularly interspaced palindromic repeats), and Cas 9, an endonuclease that determines double-stranded DNA breaks (DSBs). DSB repair may be effected through nonhomologous end-joining, in which the broken DNA ends are religated, often with additional nucleotide deletions or insertions. Another mechanism is homology-directed repair (HDR), which involves using DNA templates to accurately repair the damaged DNA. HDR is only active in dividing cells.30 CRISPR-Cas nucleases are usually used to disrupt target genes, since the indels after nonhomologous end-joining cause frameshift mutations that impede the production of a functional protein. Base editors contain a CRISPR-Cas nuclease unable to make DSBs (nickase) and a DNA deaminase enzyme and include cytosine base editors (CG- to TA) and adenine base editors (AT to GC). This technique does not require DSB or donor DNA templates, but cannot determine transversion point mutations.30 Cas transposases may integrate large genomic sequences in target sites, bit this technique has not been reported in mammalian cells.

Prime editing is a new technology that may determine all possible types of point mutations, small insertions and small deletions in a targeted way. Prime editors are composed of a Cas 9 domain fused to a reverse transcriptase and are guided to the target DNA by a prime editing guide RNA (pegRNA) that also encodes the desired edit. The reverse transcriptase copies the template from the pegRNA into the DNA that is incorporated into the DNA duplex.30 Although highly promising, this approach is limited by the dimensions of the edited DNA and by the development of adequate delivery strategies.
DELIVERY SYSTEMSSeveral viral vectors are used to effectively deliver genetic material.
LentivirusLentiviruses are enveloped single-stranded RNA viruses that are able to infect both dividing and nondividing cells.31 These viruses deliver their genetic payload by integrating it into the host cell's genome, creating a durable response within the infected cell and subsequent progeny cells.31 However, this mechanism increases the potential for insertional oncogenesis, an adverse event that has been mitigated by the development of self-inactivating lentiviral vectors.32–34
AdenovirusAdenoviruses (AVs) are nonenveloped icosahedral viruses with a double-stranded DNA genome that can hold large transgenes up to 54kb.35 The adenovirus has a high rate of gene transduction, including in cardiomyocytes, but the protein expression may be short-lived, lasting up to 4 weeks.36 Adenovirus can also elicit a significant inflammatory response, and the presence of pre-existing antibodies resulting from prior exposure to wild-type adenovirus can diminish its transduction efficiency.37
Adeno-associated virusAAV is a nonenveloped virus with a single-stranded DNA genome that is replication-deficient.36 Unlike wtAAV, recombinant AAVs (rAAV) do not integrate into the human chromosome and the viral genome remain extrachromosomal in a circular episomal form.38 Some serotypes of AAV have a natural tropism for specific tissues, such as AAV9 for the liver and heart. Although AAVs are less immunogenic than AVs, they still have the potential for immune activation and increased clearance by pre-existing neutralizing antibodies.36,39 The gene size capacity in AAV is limited to 4.7kb. Therefore, an approach using dual AAV vectors expressing different genome sequences that merge before translation is under development.40 rAAV are mostly sequestrated in the liver after systemic injection and gene delivery to specific tissues, like muscular tissue including the heart, requires high doses of the virus. Hence, AAV capsid containing specific peptides have been developed to improve transfection in muscular cells known as MyoAAV.41
Nonviral vectorsNonviral vectors have been developed to overcome the limitations of the viral vectors and include polymers, lipids, inorganic particles and combined approaches. Most of these systems are cationic and may be combined with negatively charged DNA generating an overall positive vector-gene complex that may bind to the negatively charged molecules of the cellular membrane and be internalized.42
Lipid nanoparticles (LNPs) are an attractive solution as they can be conjugated with specific targeting ligands to reach specific tissues. These nanoparticles can be modified to reduce immunogenicity, and are affordable to produce at scale.43–46 Much of the research around this novel vehicle centers around the delivery of RNA-based therapies. LNPs have been recently used in gene editing, but their effectiveness in transducing cardiomyocytes has been limited. 47,48
TOXICITY RISKS OF ADENO-ASSOCIATED VIRUS VECTORSImmune response to AAV vectors may cause several adverse events, including liver toxicity and thrombotic microangiopathy. The immunological mechanisms and potential immunosuppressive treatments are reported in table 1.
Immune response to adeno-associated virus
| Immune response to AAV | Trigger | Effect | Treatment/prevention |
|---|---|---|---|
| Pre-existing immunity | Originates from previous exposure to wtAAVNab vs wtAAV prevalence up to 70% for AAV1 and AAV2, 45% for AAV6 and AAV9, and 38% for AAV8.49 Nab seroprevalence is age-dependent, lowest value between 3 and 17 years50 | Vector neutralization | Exclusion of patients with high titer Nab |
| Innate immunity | Nonspecific response and does not generate immunological memory triggered by capsid, genome, transgene product, dsDNA, dsRNA, unmethylated CpG motifs51,52 | Transduced cell damageLoss of transgene expression | Glucocorticoids |
| Adaptive immunity | Develops after innate immunityActivation and differentiations of T and B lymphocytes and creation of memory lymphocytes that may be reactivated | Block readministrationComplement activationNeutralization of transgene productTransduced cell damageLoss of transgene expression | GlucocorticoidsRapamycinTacrolimusRituximab |
dsDNA, double-stranded; dsRNA, double-stranded RNA; Nab, neutralizing antibodies; wtAAV, wild-type adeno-associated virus.
Hepatotoxicity is the most common adverse event associated with systemic administration of AAV vectors. This adverse effect may present as elevated liver enzymes (AST, ALT), and liver failure. In clinical trials that evaluated the safety and efficacy of AAV carrying the FIX transgene to treat hemophilia B,53,54 transgene expression gradually declined after 4 to 8 weeks and was associated with a transaminase increase and the detection of AAV-specific CD8+T cells. The adverse event was limited, excluding patients with neutralizing antibodies (Nab) and improving liver expression of the transgene, and the transaminase elevation was controlled with a tapering course of steroids.54
Thrombotic microangiopathyThrombotic microangiopathy is characterized by damage to arterioles and capillaries, leading to microvascular thrombosis with clinical manifestations including hemolytic anemia, thrombocytopenia, and acute kidney injury. Complement-mediated thrombotic microangiopathy (atypical hemolytic-uremic syndrome, aHUS) is a dose-related class effect of intravenous (iv) administration of AAV for cardiac and muscle tissues. The mechanism includes rapid IgM rise, capsid binding and classic and alternative complement pathway activation. aHUS has been described in patients in gene therapy clinical trials for spinal muscular atrophy, Duchenne muscular dystrophy (DMD), Danon disease and Fabry disease (FD). In some cases, eculizumab, a monoclonal IgG antibody that prevents the cleavage of C5 into C5a and AC5b and avoiding the formation of the membrane attack complex, was used to effectively treat the condition.55,56 It is still unknown whether prophylactic use of eculizumab may be effective in preventing this adverse event.
CLINICAL TRIALS EVALUATING GENE THERAPY IN CARDIOMYOPATHIESSeveral clinical trials evaluating gene therapy in cardiomyopathies are ongoing (table 2). In this section, we describe the preliminary data from these trials, with a particular focus on gene replacement therapy using AAVs, currently the most frequently used technique.
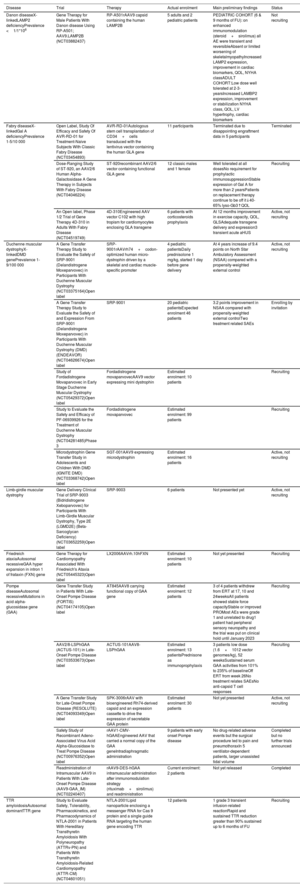
Clinical trials on gene therapies in cardiomyopathies
| Disease | Trial | Therapy | Actual enrolment | Main preliminary findings | Status |
|---|---|---|---|---|---|
| Danon diseaseX-linkedLAMP2 deficiencyPrevalence <1/1*106 | Gene Therapy for Male Patients With Danon disease Using RP-A501; AAV9.LAMP2B (NCT03882437) | RP-A501rAAV9 capsid containing the human LAMP2B | 5 adults and 2 pediatric patients | PEDIATRIC COHORT (6 & 9 months of FU): on enhanced immunomodulation (steroid+sirolimus) all AE were transient and reversibleAbsent or limited worsening of skeletalmyopathyIncreased LAMP2 expression, improvement in cardiac biomarkers, QOL, NYHA classADULT COHORT:Low dose well tolerated at 2-3-yearsIncreased LAMBP2 expression, improvement or stabilization NYHA class, QOL, LV hypertrophy, cardiac biomarkers | Not recruiting |
| Fabry diseaseX-linkedGal A deficiencyPrevalence 1-5/10 000 | Open Label, Study Of Efficacy and Safety Of AVR-RD-01 for Treatment-Naive Subjects With Classic Fabry Disease (NCT03454893) | AVR-RD-01Autologous stem cell transplantation of CD34+cells transduced with the lentivirus vector containing the human GLA gene | 11 participants | Terminated due to disappointing engraftment data in 5 participants | Terminated |
| Dose-Ranging Study of ST-920, an AAV2/6 Human Alpha-Galactosidase A Gene Therapy in Subjects With Fabry Disease (NCT04046224) | ST-920recombinant AAV2/6 vector containing functional GLA gene | 12 classic males and 1 female | Well tolerated at all dosesNo requirement for prophylactic immunosuppressionStable expression of Gal A for more than 2 yearsPatients on replacement therapy continue to be off it↓40-65% lyso-Gb3↑QOL | Recruiting | |
| An Open label, Phase 1/2 Trial of Gene Therapy 4D-310 in Adults With Fabry Disease (NCT04519749) | 4D-310Engineered AAV vector C102 with high tropism for cardiomyocytes enclosing GLA transgene | 6 patients with corticosteroids prophylaxis | At 12 months improvement in exercise capacity, QOL, GLSAdequate transgene delivery and expression3 transient acute aHUS | Active, not recruiting | |
| Duchenne muscular dystrophyX-linkedDMD genePrevalence 1-9/100 000 | A Gene Transfer Therapy Study to Evaluate the Safety of SRP-9001 (Delandistrogene Moxeparvovec) in Participants With Duchenne Muscular Dystrophy (NCT03375164)Open label | SRP-9001rAAVrh74+codon-optimized human micro-dystrophin driven by a skeletal and cardiac muscle-specific promoter | 4 pediatric patientsDaily prednisolone 1 mg/kg, started 1 day before gene delivery | At 4 years increase of 9.4 points on North Star Ambulatory Assessment (NSAA) compared with a propensity-weighted external control | Active, not recruiting |
| A Gene Transfer Therapy Study to Evaluate the Safety of and Expression From SRP-9001 (Delandistrogene Moxeparvovec) in Participants With Duchenne Muscular Dystrophy (DMD) (ENDEAVOR) (NCT04626674)Open label | SRP-9001 | 20 pediatric patientsExpected enrolment 46 patients | 3.2 points improvement in NSAA compared with propensity-weighted external controlTwo treatment related SAEs | Enrolling by invitation | |
| Study of Fordadistrogene Movaparvovec in Early Stage Duchenne Muscular Dystrophy (NCT05429372)Open label | Fordadistrogene movaparvovecAAV9 vector expressing mini dystrophin | Estimated enrolment: 10 patients | Recruiting | ||
| Study to Evaluate the Safety and Efficacy of PF-06939926 for the Treatment of Duchenne Muscular Dystrophy (NCT04281485)Phase 3 | Fordadistrogene movaparvovec | Estimated enrolment: 99 patients | Recruiting | ||
| Microdystrophin Gene Transfer Study in Adolescents and Children With DMD (IGNITE DMD) (NCT03368742)Open label | SGT-001AAV9 expressing microdystrophin | Estimated enrolment: 16 patients | Active, not recruiting | ||
| Limb-girdle muscular dystrophy | Gene Delivery Clinical Trial of SRP-9003 (Bidridistrogene Xeboparvovec) for Participants With Limb-Girdle Muscular Dystrophy, Type 2E (LGMD2E) (Beta-Sarcoglycan Deficiency)(NCT03652259)Open label | SRP-9003 | 6 patients | Not presented yet | Active, not recruiting |
| Friedreich ataxiaAutosomal recessiveGAA hyper expansion in intron 1 of frataxin (FXN) gene | Gene Therapy for Cardiomyopathy Associated With Friedreich's Ataxia (NCT05445323)Open label | LX2006AAVrh.10hFXN | Estimated enrolment: 10 patients | Not yet presented | Recruiting |
| Pompe diseaseAutosomal recessiveMutations in acid alpha-glucosidase gene (GAA) | Gene Transfer Study in Patients With Late-Onset Pompe Disease (FORTIS) (NCT04174105)Open label | AT845AAV8 carrying functional copy of GAA gene | Estimated enrolment: 12 patients | 3 of 4 patients withdrew from ERT at 17, 10 and 24weeksAll patients showed stable force capacityStable or improved PROMost AEs were grade 1 and unrelated to drug1 patient had peripheral sensory neuropathy and the trial was put on clinical hold until January 2023 | Recruiting |
| AAV2/8-LSPhGAA (ACTUS-101) in Late-Onset Pompe Disease (NCT03533673)Open label | ACTUS-101AAV8-LSPhGAA | Estimated enrolment: 13 patientsPrednisone as immunoprophylaxis | 3 patients low dose (1.6×1012 vector genomes/kg), 52 weeksSustained serum GAA activities from 101% to 235% of baselineOff ERT from week 26No treatment relates SAEsNo anti-capsid T cell responses | Recruiting | |
| A Gene Transfer Study for Late-Onset Pompe Disease (RESOLUTE) (NCT04093349)Open label | SPK-3006rAAV with bioengineered Rh74-derived capsid and an expression cassette to drive the expression of secretable GAA protein | Estimated enrolment: 30 patients | Not yet presented | Active, not recruiting | |
| Safety Study of Recombinant Adeno-Associated Virus Acid Alpha-Glucosidase to Treat Pompe Disease (NCT00976352)Open label | rAAV1-CMV-hGAAEngineered AAV that carried a normal copy of the GAA geneIntradiaphragmatic administration | 9 patients with early onset Pompe disease | No drug-related adverse events but the surgical procedure led to pain and pneumothoraxIn 5 ventilator-dependent patients, larger unassisted tidal volume | Completed but no further trials announced | |
| Readministration of Intramuscular AAV9 in Patients With Late-Onset Pompe Disease (AAV9-GAA_IM) (NCT02240407) | rAAV9-DES-hGAA intramuscular administration after immunomodulation strategy (rituximab+sirolimus) and readministration | Current enrolment: 2 patients | Not yet released | Completed | |
| TTR amyloidosisAutosomal dominantTTR gene | Study to Evaluate Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of NTLA-2001 in Patients With Hereditary Transthyretin Amyloidosis With Polyneuropathy (ATTRv-PN) and Patients With Transthyretin Amyloidosis-Related Cardiomyopathy (ATTR-CM) (NCT04601051) | NTLA-2001Lipid nanoparticle enclosing a messenger RNA for Cas 9 protein and a single guide RNA targeting the human gene encoding TTR | 12 patients | 1 grade 3 transient infusion-related reactionRapid and sustained TTR reduction greater than 90% sustained up to 6 months of FU | Recruiting |
AAV, adeno-associated virus; AE, adverse events; AHUS, atypical hemolytic uremic syndrome; DMD, Duchenne muscular dystrophy; ERT, enzyme replacement therapy, FU, follow-up; GAA, acid alpha-glucosidase gene; Gal A, alpha-galactosidase A; GLA, galactosidase alpha gene; GLS, global longitudinal strain; LAMP, lysosomal associated membrane protein-2; LAMP2B, lysosome-associated membrane protein 2 isoform B; Lyso-Gb3, globotriaosylsphingosine; LV, left ventricle/ventricular; NYHA, New York Heart Association; NSAA, North Star Ambulatory Assessment; PRO, patient reported outcome; QOL, quality of life; rAAV, recombinant adeno-associated; TTR, transthyretin.
Danon disease is a rare X-linked lysosomal disorder due to pathogenic variants in the LAMP2 (lysosomal associated membrane protein 2) gene causing impaired autophagy and the accumulation of autophagic vacuoles in muscle tissue. Clinical manifestations include rapidly progressive hypertrophic cardiomyopathy (HCM) with a median survival for males of 19 years without heart transplantation.57 No disease-modifying pharmacological therapies are available for this condition. A single iv infusion of RP-A501, an AAV9 vector expressing LAMP2B, at a low dose (6.7 x 10^13 GC/kg) or high dose (1.1 x 10^14 GC/kg) in male patients aged 8 to 14 years and ≥ 15 years has been evaluated in an open-label phase 1 study.58 The preliminary results of the study were presented at the 2022 Heart Failure Society of America annual scientific meeting. In 3 patients in cohort 1 (≥ 15 years, low dose), RP-A501 increased LAMP2B expression by Western blot and immunochemistry in myocardial tissue. The 2 patients with observed immunosuppressive regime compliance showed high cardiac LAMP2B expression (67.8% and 92.4% vs normal control by immunochemistry) and improvement in New York Heart Association (NYHA) class and natriuretic peptides. All 3 patients showed improvement or stability of functional capacity measured by the 6-minute walk test, an increase in physical activity, and decreased autophagic vacuoles on endomyocardial biopsy. The most common adverse events included reversible platelet decrease, transaminase elevation, and steroid-induced myopathy. In the high-dose adult cohort, 1 patient developed complement-mediated thrombotic microangiopathy with thrombocytopenia and acute kidney injury requiring transient hemodialysis. Consequently, no further patients have been enrolled in the higher dose cohort; furthermore, the immunosuppressive regimen was adjusted, limiting the daily dose of corticosteroids, using sirolimus instead of tacrolimus to minimize the renal impact and continuing rituximab. In September 2022, an update of the phase 1 trial was presented.59 In the adult cohort, a significant reduction in biomarkers, reduced left ventricular wall thickness, improvement or stability of NYHA class and Kansas city cardiomyopathy questionnaire score were sustained up to 36 months of follow-up. In the 2 patients in the pediatric cohort, with the improved immunomodulation protocol, there were no reports of thrombocytopenia, skeletal myopathy, late transaminitis or complement activation-related adverse events. Protein expression was improved at the latest follow-up at 3 and 6 months. Moreover, an improvement in biomarkers, reported symptoms and NYHA class was seen at 6 and 9 months. LAMP2 expression was increased similar to the adult cohort at the same timeframe and the vacuolar area at the available endomyocardial biopsy was significantly reduced. On the basis of these positive results, the United States FDA has granted Regenerative Medicine Advanced Therapy designation to RP-A501, and phase 2 trial is scheduled to start in the second quarter of 2023.60
Fabry diseaseFD is an X-linked lysosomal storage disorder due to pathogenic variants in the GLA (galactosidase alpha) gene. This alteration leads to various degrees of alpha-galactosidase (Gal A) deficiency and accumulation of glycosphingolipids, mostly globotriaosylceramide (Gb3) and its deacylated derivative globotriaosylsphingosine (lyso-Gb3), in target organs such as the heart, kidney and blood vessels. The current standard of care includes iv enzyme replacement therapy or oral chaperone therapy, which, although effective in ameliorating quality of life, neuropathic pain, gastrointestinal manifestation and renal function, has limited efficacy in cardiomyopathy.61
FD is a suitable candidate for gene therapy as it is a monogenic disease and clinical effects can be achieved with 10% activity of normal Gal A levels. Several studies are currently evaluating the safety and efficacy of gene therapy in FD.
AVR-RD-01In an open-label study assessing AVR-RD-01, autologous stem cell transplantation of CD34+cells transduced with a lentivirus vector containing the human GLA gene was used. However, recruitment for the trial has been halted due to disappointing engraftment data in 5 participants.62
ST-920By means of ST-920 (isaralgagene civaparvovec), functional GLA gene is delivered to the liver via one-time infusion of a recombinant AAV2/6 vector without the need for immunosuppressive therapy. This approach is currently under study in the phase 1/2 STAAR trial (NCT04046224). In the dose escalation phase of the trial, 9 male patients with classic FD with mild/moderate disease severity were enrolled. In the expansion phase, 5 patients received a dose of 5 x 10^13 vg/kg. As of the October 2022 data cutoff date, no treatment-related adverse events greater than grade 2 have been reported in the 13 patients studied.
All 9 patients treated in the dose escalation phase across the 4 dose cohorts (0.5 x 10^13 vg/kg, 1 x 10^13 vg/kg, 3 x 10^13 vg/kg, and 5 x 10^13 vg/kg) showed dose-dependent, supraphysiological and sustained Gal A activity ranging from nearly 4-fold to 68-fold of the mean normal levels for more than 2 years for the patient with the longest follow-up. A stable and dose-dependent reduction in Lysogb3 concentration was observed at high baseline levels (> 80 ng/mL) and, at the highest dose, also at low plasma levels (< 25 ng/mL) up to 25 months of follow-up. The therapy was also associated with a potential improvement in Fabry nephropathy. Indeed, a reduction in Gb3 inclusions and podocyte loss has been reported in patient 9.
C102C102, an engineered AAV vector with high tropism for cardiomyocytes, was found to transduce nonhuman primate heart tissue in vivo along with human cardiomyocytes and endothelial cells in vitro better than AAV1, AAV8 or AAV9-serotyped vectors, allowing the use of low dose iv delivery.63
4D-310 (vector C102 enclosing GLA transgene) promotes high and stable blood Gal A levels that can be taken up by cells in the proximity, but also permits transduction directly to target tissue cells such as cardiomyocytes, kidney (glomeruli including podocytes), and blood vessels.64
The phase 1/2 open-label clinical trial INGLAXA-1 and -2 is currently assessing the safety and tolerability of 4D-310 in patients with classic or late-onset FD with cardiac involvement on or off enzyme replacement therapy with corticosteroid prophylactic immunosuppression. In cohort 1 (1^13 vg/kg with corticosteroid immune regimen), 6 patients have been enrolled, 4 had enzyme replacement therapy experience and 1 was on chaperone therapy. Global longitudinal strain and peak VO2 improved from baseline to week 52 in 2 of 3 patients. One patient underwent cardiac biopsy in week 6, and the tissue proved healthy with no signs of inflammation. In situ hybridization showed uptake in cardiomyocytes only, with around 50% of cardiomyocytes positive for transgene expression. On Immunohistochemistry, AGA protein was expressed in all samples. Genome delivery was adequate, being 4.4 vector copies per cardiomyocyte and RNA expression was 16.2 transcript copies per cardiomyocyte.
Three patients experienced transient acute aHUS, with onset at day 3 to 7; 1 patient required no intervention, 1 was treated with eculizumab, and 1 was treated with eculizumab and temporary dialysis. After the aHUS dose limiting toxicity, the FDA put the program on clinical hold.
The patients’ immune response was therefore thoroughly assessed and the patients developed aHUS showed predosing complement activation. The next steps for this trial include alignment with the FDA to remove the clinical hold and resume enrolment following protocol amendment including implementation of a rituximab/sirolimus immunosuppressive regimen, and adding prescreening for complement and excluding patients with predosing complement activation.
Duchenne muscular dystrophyThe large size of the DMD gene poses challenges for the potential application of gene transfer therapies. However, a smaller gene construct may be able to alleviate the disease phenotype and several recombinant genes encoding mini/micro-dystrophin have been created.65
A phase 3 clinical trial on fordadistrogene movaparvovec (SRP-9001), an AAV9 vector expressing mini-dystrophin, is ongoing after an initial clinical hold from the FDA due to the death of a participant (NCT04281485).66
SRP-9001 is formed by rAAV rh74 (rAAVrh74) and a codon-optimized human micro-dystrophin driven by a skeletal and cardiac muscle-specific promoter with enhanced cardiac expression. 67 Preliminary results of the phase 1/2a nonrandomized controlled trial have shown an improvement in functional motor abilities in 4 pediatric patients after 4 years and a phase 3 trial is ongoing.68
The exon skipping technique allows silencing of specific exons determining the restoration of the reading frame of a mutated gene by means of silencing RNA, antisense oligonucleotides or gene editing. Exon skipping is a mutation-specific technique and, in DMD it leads to the production of shortened but functional dystrophin protein. Four antisense oligonucleotide therapies have been approved by the FDA for DMD treatment and an AAV vector delivering small nuclear RNA that contains antisense sequences targeting the duplicated DMD exon 2 is under study.65
Friedreich ataxiaA phase 1A/1B study is currently evaluating the safety and efficacy of a serotype rh.10 AAV delivering a functional frataxin (FXN) gene for the treatment of Friedreich ataxia cardiomyopathy (NCT05445323). In a murine model, this approach reversed cardiac mass, improved cardiac function, corrected FXN-related biochemical defects and reversed stress-induced cardiac dysfunction.69,70
Ribonucleoprotein complex for the excision of the acid alpha-glucosidase (GAA) repeat sequences in FXN has shown promising results in a murine model and a clinical trial is in the planning phase.71
Pompe diseaseSeveral gene therapies designed to provide a functional copy of the GAA gene are at various stages of development.
In 2015, the first-in-human trial of diaphragmatic gene therapy (AAV1-CMV-GAA) to treat respiratory and neural dysfunction in early-onset Pompe disease was conducted. No adverse events related to the drug have been reported, but intradiaphragmatic injection led to pneumothorax and lung contusion, and ventilatory performance was only modestly improved.
The initial analysis of 3 patients with late-onset Pompe disease in the trial of ACTUS-101, and AAV8 vector expressing human GAA under the transcriptional control of a liver-specific promoter (AAV2/8-LSPhGAA), showed a sustained increase in GAA levels and no treatment-related SAEs.72
AT845 is an AAV8 designed to express human GAA specifically in skeletal muscle and heart and is currently under study in patients with late-onset Pompe disease with promising preliminary results.73
TRANSTHYRETIN AMYLOIDOSISThe CRISPR-Cas9 system is under evaluation for the treatment of transthyretin (TTR) amyloidosis. TTR amyloidosis is characterized by progressive deposition of misfolded senescent or mutated transthyretin in various tissues including the heart and peripheral nervous system. NTLA-2001 is included in an LNP opsonized by apolipoprotein E that acts as a carrier system. The active components are a messenger RNA for a Cas 9 protein and a single guide RNA (sgRNA) targeting the human gene encoding TTR. NTLA-2001 LNP is administered iv and reaches the liver where is taken up by low-density lipoprotein receptor expressed by hepatocytes. The Cas9 RNA molecule is translated in the cytoplasm, producing the Cas9 endonuclease enzyme. Along with the Cas9 endonuclease, the TTR-specific sgRNA forms a CRISPR-Cas9 ribonucleoprotein complex. The latter enters the nucleus, the sgRNA binds the target DNA, and the CRISPR-Cas9 complex may unwind the helix and reach the target gene. The TTR gene is eventually cleaved and endogenous DNA repair through nonhomologous end-joining introduces insertions or deletions of bases (indels) that result in the reduction in the levels of functional mRNA and transthyretin. In human hepatocytes, NTLA-2001 produced saturating levels of TTR editing (≥ 93.7%), resulting in a reduction of at least 91% of TTR mRNA expression and TTR protein. Next-generation sequencing showed that NTLA-2001 induced TTR knock-out. Preclinical studies in transgenic mice and cynomolgus monkeys showed that TTR editing led to a sustained reduction in TTR levels over a 12-month period. An open-label, single dose, proof-of-concept study has enrolled 6 patients, aged 46 to 64 years, with hereditary TTR amyloidosis polyneuropathy. All patients completed the infusion and 1 patient had a grade 1 infusion-related reaction. Coagulation values, platelet count and transaminase levels remained within the reference ranges. A rapid and sustained dose-dependent reduction in TTR concentration was observed, with a reduction at day 28 between 47% and 56% in the lower dose group (0.1mg/kg) and between 80% and 96% in the higher dose group (0.3mg/kg).47
Data from the 12 patients with hereditary or wild-type cardiomyopathy in the dose escalation phase (9 patients on 0.7mg/kg, 3 patients on 1.0mg/kg) have been presented. NTLA-2001 was well tolerated and there was a single grade 3 infusion-related reaction, which resolved without clinical sequalae. All patients achieved a TTR concentration reduction greater than 90% by day 28 that was sustained throughout the 6 months of follow-up.74
FUTURE TARGETSMYH7Preclinical models for CRISPR-Cas9 gene editing are also under study for HCM. The latter is defined by a left ventricular thickness greater than 15mm and the disease may be caused by pathogenic variants in genes encoding sarcomeric proteins. The main clinical manifestations include reduced exercise capacity, heart failure, and arrhythmias. Several approaches have been proposed. Chai et al.75 used a dual AAV9 delivery system enclosing an adenine base editing (ABEmax-VRQR, which mediates the conversion of A•T to G•C without DNA cleavage) and a guide RNA (h403_sgRNA) to target the pathogenic missense variant in the MYH7 gene p.Arg403Gln. The treatment corrected the target pathogenic adenine at 98% efficiency in induced pluripotent stem cells derived from patients with HCM with minimal bystander editing of nearby adenines and low DNA off-target editing. The differentiated cardiomyocytes had a normal contractile force and cellular energetics compared with uncorrected cardiomyocytes. In a humanized mouse model of HCM, intrathoracic injection of a single dose of dual AAV9 encoding ABEmax-VRQR and the h403_sgRNA resulted in correction of the pathogenic allele, a reduction in pathogenic transcripts, and a reduction in cardiac hypertrophy and histopathologic remodeling.
Reichart et al.40 used a similar dual AAV-based strategy to deliver an ABE and gRNA to correct the same pathogenic variant in a nonhumanized mice model, obtaining a correction of the pathogenic variant in ≥ 70% of ventricular cardiomyocytes and preventing the development of the pathological phenotype. As an alternative approach, AAV9 delivery of RNA-guided Cas9 nuclease was tested. The technique effectively inactivated the pathogenic allele, but there was also reduced contractile function with higher doses, demonstrating accidental editing of the wild-type allele.
MYBPC3Loss-of-function mutations in myosin-binding protein C3 (MYBPC3) together with MYH7 are the most common genetic causes of HCM. Reduced MYBPC3 in the sarcomere eventually leads to an increase in myosin heads available for actin interaction and hypercontractility.
A single administration of AAV9-MYBPC3 in homozygous MYBPC3-targeted knock-in mice mimicking a neonatal form of hypertrophic cardiomyopathy prevented the development of cardiac hypertrophy through 34 weeks of observation, and, in a dose-dependent manner, increased MYBPC3 mRNA and protein. Furthermore, the therapy unexpectedly reduced the pathological mRNA species concentration. Probably, since the transgene MYBPC protein would be more stably integrated in the sarcomere, a feedback mechanism may downregulate the transcription of the mutant protein.76 In a further study, since the MYBPC3 N-terminal domain is involved in the interactions with actin and myosin, the transfer of the sole N-terminal domain of MYBPC through AAV9 compared with full-length has been evaluated in a cMyBPC-null murine model (cMyBPC–/–). The transfer of the N-terminal domain was sufficient to prevent the macroscopic (left ventricular hypertrophy) and microscopic (myocardial disarray and fibrosis) HCM phenotype. However, it is still unknown whether AAV therapy may rescue the HCM phenotype and induce reverse remodeling in overt HCM.77 A phase 1 clinical trial to evaluate the safety, tolerability and clinical efficacy of a single iv infusion of an AAV gene therapy for the treatment of MYBPC3 mutation-related HCM is scheduled to begin in the third quarter of 2023.78
BAG3The BAG3 (BCL2-associated athanogene 3) gene is highly expressed in the heart, skeletal muscle, and central nervous system. BAG3 in cardiomyocytes enhances contractility by linking the beta-adrenergic receptor and L-type Ca2+channel, facilitates autophagy interacting with heat shock proteins, provides support for the sarcomere by linking actin with the Z- disc, and has antiapoptotic properties through BCL2 binding.79 Truncating or deletion variants in the BAG3 gene causing reduced levels of BAG3 protein have been associated with dilated cardiomyopathy (DCM).80 Human DCM cardiomyocytes have reduced sarcomere contractile function and impaired myofilament turnover with misfolded/dysfunctional proteins that remain integrated in the sarcomere. Moreover, myofilament expression of BAG3 decreases in DCM and is correlated with reduced contractile function. In mice receiving a myocardial infarction, the administration of recombinant AAV9 vector expressing mouse BAG3 gene restored cardiac function and myofilament turnover.81 Furthermore, iv delivery of adeno-associated virus 9 (AAV9)-BAG3 in Bag3 haplo-insufficient mice prevented the onset of reduced ejection fraction.82
In a recent report, catheter-based retrograde coronary sinus infusion was safely used to deliver low doses of AAV9-BAG3 in healthy mini pigs, resulting in diffuse transduction in the myocardium.83 A phase 1/2 clinical trial to evaluate the safety and efficacy of a single administration via retrograde coronary sinus infusion in patients with reduced ejection fraction and BAG 3 haploinsufficiency is the planning phase.60
PKP2Arrhythmogenic cardiomyopathy is a myocardial disease characterized by fibrofatty replacement of the right or left ventricle, or both. The structural modifications create a proarrhythmic milieu and determine progressive chamber dilation and systolic dysfunction. Arrhythmogenic cardiomyopathy in 40% to 50% of cases is associated with mutations in desmosomal genes, with plakophilin-2 (PKP2) being the most frequent. An AAVrh10 vector expressing the PKP2 gene (LX2020) has been evaluated in PKP2 mutant mice. LX2020 showed a dose-dependent improvement in survival of the mice, a reduction in ectopic beats, and amelioration of left and right ejection fraction.84 Preclinical data on PKP2 knock-out mice have also been reported for TN-401 (AAV9 vector expressing PKP2), showing a reduction in ventricular arrhythmias, prevention of right and left ventricular dysfunction, and extension of the life span. Furthermore, the therapy proved effective in restoring desmosomes and gap junctions and in preventing fibrosis.85 Clinical trials evaluating these strategies are in the design phase.
PRKAG2PRKAG2 mutations lead to a glycogen storage cardiomyopathy characterized by hypertrophic phenotype, supraventricular arrhythmias and atrioventricular blocks. A CRISPR-Cas9 system combined with AAV9 has been used to disrupt the mutant PRKAG2 allele encoding the H503 mutation in knock-in mice. A single systemic injection of the product at day 4 or day 42 restored the morphology and function in the mice model, reducing left ventricular thickness and myocardial glycogen content.86
LMNAHutchinson-Gilford progeria syndrome is characterized by premature aging and death due to cardiovascular causes and is provoked by mutations in the LMNA gene that is translated into progerin, exerting a dominant-negative effect. CRISPR-Cas9 technology has been used to generate mice that ubiquitously express progerin and lamin C and lack lamin A and allows progerin suppression and lamin A restoration in a time- and cell-type-specific manner on Cre recombinase activation. Progerin suppression and lamin A restoration restricted to vascular smooth cells and cardiomyocytes prevented vascular damage and normalized the lifespan in this murine model.87
CURRENT CHALLENGESGene therapy could be a real game changer for different diseases; however, several issues need to be resolved. The greatest challenges with AAV gene therapy include improving transduction efficacy, finding the best strategy to target specific tissues, reducing the administered doses to minimize possible adverse events, identifying the most effective immunosuppressive strategies to blunt the immune response and eventually allow a readministration of the drug.
Gene editing systems may have off-target activity, namely altering the genome in regions other than the target site. High fidelity Cas variants can reduce off-target editing but are also associated with lower on-target activity.30
Although several options have been developed, a tissue-specific, nonimmunogenic adequate delivery system remains an unmet need.
Patient selection remains unsolved; indeed, it is still unknown whether the treatment could rescue an already overt phenotype or if it should be administered in the early phase of the disease.
Long-term durability and long-term effects are still unknown.
Further problems include affordability, regulation, and access to the therapy. All these approaches have high manufacturing costs and access to therapy may be limited.
CONCLUSIONGene therapy is an evolving and promising field that already allows targeted treatment for severe diseases like Leber congenital amaurosis and spinal muscular atrophy. Safety and durability issues remain to be resolved; however, several ongoing clinical trials will hopefully provide answers and expand the technique to other conditions.
FUNDINGThe authors received no specific funding for this work.
AUTHORS’ CONTRIBUTIONSE. Adler contributed in the work through the activities of conceptualization, supervision, review, and editing. A. Argirò and J. Ding wrote the original draft and made edits under E. Adler's supervision.
CONFLICTS OF INTERESTE. Adler participates in the Scientific Advisory Board for Medtronic, Fuji, and SanaChief. E. Adler works as scientific officer for Lexeo Therapeutics, and as a consultant for Abiomed, AstraZeneca, Ionis, Medtronic, Abbott, and Novartis. E. Adler is part of the advisory board and shareholder for Rocket Pharmaceuticals. E. Adler is a founder of the scientific board and shareholder of ResQue Therapeutics. A. Argirò and J. Ding have no conflicts of interest to declare.
.