Sra. Editora:
El síndrome de Holt-Oram (SHO) asocia anomalías esqueléticas de las extremidades superiores con alteraciones cardiovasculares. Para realizar el diagnóstico de SHO deben estar presentes las alteraciones esqueléticas características y posteriormente se demostrará la afección cardiaca, en el mismo individuo o en sus progenitores, mostrando la evidencia de su transmisión genética1.
Se describen anomalías esqueléticas en las extremidades superiores del 100% de los afectados, con mayor frecuencia de los pulgares. El aparato cardiovascular se encuentra afectado en el 75% de los diagnosticados, lo más característico es la comunicación interauricular. Es conocida la ausencia de correlación entre la gravedad de las lesiones óseas y cardiacas, así como lo heterogéneo en los familiares afectados.
En 1997 fue descubierto el gen causal, TBX5 en el cromosoma 12. La transmisión es autosómica dominante con un 100% de penetrancia. La localización y el tipo de mutación en TBX5 no son predictores de la expresividad fenotípica. La probabilidad de encontrar la mutación en el gen TBX5 es del 74% en los pacientes que cumplen los criterios clínicos de SHO2.
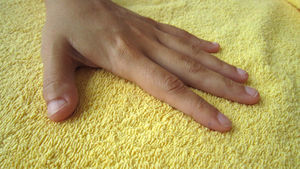
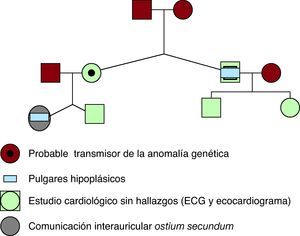
El caso clínico que presentamos es una mujer de 14 años diagnosticada de una comunicación interauricular tipo ostium secundum, junto con dedos pulgares hipoplásicos (Figura 1). Un tío (materno) tiene similar malformación de pulgares. El estudio cardiológico de todos los candidatos a heredar la mutación (Figura 2), consistente en ecocardiograma Doppler y electrocardiograma, no encontró anomalías. A la madre, al considerarla probable transmisora del trastorno genético y sin repercusión ósea ni cardiaca, se le ha realizado radiografía de manos, que descarta la existencia de malformación ósea subclínica.

Figura 1. Pulgar hipoplásico. Desproporción de la falange distal.

Figura 2. Genograma familiar. La madre de la paciente es probable portadora de la anomalía genética sin manifestaciones fenotípicas en pulgares, ni radiológicas subclínicas en huesos del carpo.
Obtenido el consentimiento informado, se ha extraído y purificado el ADN genómico de la paciente y, tras amplificación específica mediante PCR de todos los exones y regiones intrónicas flanqueantes del gen TBX5, se ha procedido a la secuenciación directa en doble cadena de todos los fragmentos de PCR purificados. No se ha encontrado ninguna variación en la secuencia nucleotídica que permita confirmar molecularmente el SHO.
Borozdin et al. en 2006 detectaron deleción de uno o varios exones, o incluso del gen TBX5 completo en cerca de un 2% de individuos con SHO que no presentaban mutaciones identificadas mediante secuenciación3. El estudio de esta familia se ha completado mediante la realización de MLPA (Multiplex Ligation-dependent Probe Amplification) con el kit P180-B1 (MRC-Holland, Amsterdam, Países Bajos) y se han descartado también duplicaciones y/o deleciones en los 10 exones del gen TBX5 y se ha comprobado la existencia de dos copias de este.
La mutación de novo o el mosaicismo se descarta al tener un antecedente familiar de primer grado afecto de la alteración ósea característica. La ausencia de otras alteraciones (sordera, anemia, renales, etc.) elimina otros síndromes (Okihiro y acrorrenoocular), habitualmente debidos a mutación de SALL4. Dentro de las alteraciones relacionadas con el gen SALL4, se ha descrito excepcionalmente una presentación de SHO clásica4. La baja prevalencia de estas malformaciones hace excepcional una coincidencia accidental. Consideramos a la familia afectada por un SHO, del 25% en las que no se detecta la anomalía genética clásica.
Destacamos dos aspectos clínicos relevantes respecto a lo descrito por la literatura: la afección ósea es similar en los pacientes afectados y la probable transmisora (madre de la paciente) no presenta manifestaciones óseas ni cardiacas, lo cual es excepcional en las familias reportadas.
En las anomalías del gen SALL4 está descrita la penetrancia no total, miembros de dos familias con mutación identificada y sin afección clínica. Esta penetrancia no total no se ha descrito en el SHO asociado al TBX5 ni en familias con SHO clínico.
Agradecimientos al Instituto de Salud Carlos III por el contrato de Técnico Superior de Apoyo a la Investigación concedido.
Autor para correspondencia: nmurga@telefonica.net