To the Editor,
Holt-Oram syndrome (HOS) is a disorder characterized by skeletal abnormalities in the upper limbs accompanied by cardiovascular defects. The diagnosis of HOS is based on these abnormalities and defects, both in the individual or in the parents, and is indicative of congenital transmission.1.
All those affected by this syndrome have upper limb skeletal abnormalities, which preferentially affect the thumbs. Of those diagnosed with this syndrome, 75% have cardiovascular defects, atrial septal defect being the most frequent. The lack of correlation between the severity of the skeletal and cardiac lesions is well known, as well as the heterogeneity of the syndrome in the affected relatives.
The causative gene TBX5, located on chromosome 12, was discovered in 1997. This has an autosomal dominant transmission with 100% penetrance. The location and type of mutation in TBX5 are not predictive of phenotypic expression. The probability of finding the mutation in the TBX5 gene is 74% in patients who fulfill the clinical criteria for HOS.2.
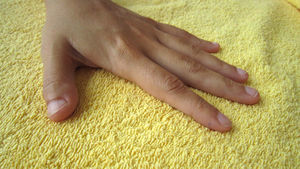
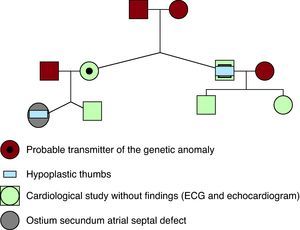
We present the clinical case of a 14-year-old girl with hypoplastic thumbs who had been diagnosed with an ostium secundum atrial septal defect (Figure 1). A maternal uncle has a similar thumb malformation. All those who were candidate for inheriting the mutation (Figure 2) underwent Doppler echocardiography and electrocardiography study with no findings of note. As the mother was considered the probable transmitter of the genetic disorder, despite having no bone or cardiac defects, an x-ray of her hands was performed which ruled out any subclinical bone malformation.

Figure 1. Hypoplastic thumb. Malformed distal phalanx.

Figure 2. Family genogram. The probable carrier of the genetic anomaly is the patient's mother, who does not present phenotypic manifestations involving the thumbs or subclinical radiological findings suggestive of carpal bone abnormalities.
After obtaining informed consent, genomic DNA from the patient was extracted and purified, followed by PCR amplification specific to all the exons and intron flanking regions of the TBX5 gene. Finally, direct sequencing of all the purified double-stranded PCR fragments was performed. No variations in the nucleotide sequence were found such that HOS could be confirmed.
In 2006, Borozdin et al. detected deletions in one or several exons or even of the entire TBX5 gene in around 2% of individuals with HOS who did not present mutations identified via sequencing.3 The study of this family was completed by MLPA (Multiplex Ligation-dependent Probe Amplification) using the P180-B1 kit (MRC-Holland, Amsterdam, the Netherlands); duplications or deletions in the 10 exons of the TBX5 gene were also ruled out and the existence of two copies of the gene checked.
De novo mutations or mosaicism were eliminated due to the presence of a first-degree relative affected by characteristic skeletal alterations. The absence of other abnormalities (deafness, anemia, renal, etc.) ruled out other syndromes (Okihiro and acro-renal-ocular) which are typically due to SALL4 mutations. Within the range of abnormalities related to the SALL4 gene, an exceptional case of classic HOS has been described.4 The low prevalence of these malformations makes such a coincidence exceptional. We consider that 25% of the family in whom the classic genetic anomaly was not detected were affected by HOS.
We draw attention to two relevant clinical aspects related to reports in the literature: the bone disorder is similar in the affected patients, and the probable transmitter (the patient's mother) has no bone or cardiac manifestations, which is exceptional in the families described in the literature.
In relation to SALL4 gene abnormalities, incomplete penetrance and an identified mutation without clinical manifestations in members of 2 families have been described. This incomplete penetrance has not been described in TBX5-associated HOS or in families with clinical HOS.
Acknowledgements
We would like to thank the Instituto de Salud Carlos III for the Research Support Engineer contract.
.
Corresponding author: nmurga@telefonica.net