
El fosfolambán es un inhibidor de la bomba de calcio sarcoplásmica que regula la contractilidad y la relajación. La mutaciones de su gen, PLN, se han asociado a fenotipos agresivos tanto de miocardiopatía dilatada como de miocardiopatía arritmogénica ventricular derecha1,2.
Se amplía la información aquí de una familia con diagnóstico de miocardiopatía arritmogénica con algunas características peculiares, portadora de una mutación fundadora holandesa en el fosfolambán (PLN c.40_42delAGA; p.Arg14del)3. La correlación genotipo-fenotipo permitió la identificación de algunas señales de alerta que deben hacer sospechar esta mutación en el estudio diagnóstico clínico.
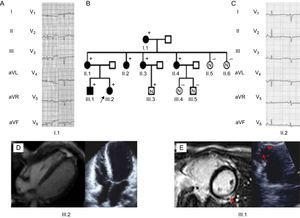
El probando (III.2) (figura 1) es una mujer de 28 años, con antecedentes previos de presíncopes. El electrocardiograma (ECG) mostró QS en derivaciones inferiores y unos voltajes notablemente bajos en todas las derivaciones (figura 2).
; + indica portadores. Las figuras negras corresponden a portadores afectados. D: la ecocardiografía y la resonancia magnética del probando mostraron volúmenes biventriculares normales, disfunción biventricular grave y ausencia de captación tardía de gadolinio. E: imágenes cardíacas y ecocardiográficas del III.1, las flechas rojas indican la mancha de captación tardía de gadolinio lateral en el ventrículo izquierdo. A la izquierda se observa un aneurisma ventricular derecho de la pared libre.")
A y C: elecrocardiograma de los portadores, los voltajes bajos y la mala progresión de la onda R son la característica distintiva. Obsérvense las ondas T invertidas en las derivaciones precordiales izquierdas. B: árbol genealógico, la flecha negra indica el probando (III.2); + indica portadores. Las figuras negras corresponden a portadores afectados. D: la ecocardiografía y la resonancia magnética del probando mostraron volúmenes biventriculares normales, disfunción biventricular grave y ausencia de captación tardía de gadolinio. E: imágenes cardíacas y ecocardiográficas del III.1, las flechas rojas indican la mancha de captación tardía de gadolinio lateral en el ventrículo izquierdo. A la izquierda se observa un aneurisma ventricular derecho de la pared libre.

La ecocardiografía mostró un ventrículo izquierdo no dilatado, con hipocinesia global y fracción de eyección del ventrículo izquierdo del 40%. La resonancia magnética cardiaca con gadolinio no mostró captación de contraste tardía. El ventrículo derecho no estaba dilatado y tenía una función sistólica global normal (fracción de eyección del ventrículo derecho del 51%); el vértice era notablemente hipocinético.
Los resultados de los análisis de sangre fueron anodinos. La tomografía computarizada coronaria mostró unas coronarias normales. El registro Holter de 24 h fue normal. Se dio el alta a la paciente en tratamiento con bloqueadores beta, un inhibidor de la enzima de conversión de la angiotensina y espironolactona.
Reingresó 3 meses después por un nuevo colapso. Durante ese ingreso, se documentó una taquicardia ventricular sostenida (figura 2) que motivó implante de desfibrilador automático implantable. La paciente sigue en clase funcional I de la New York Heart Association (NYHA). El análisis genético descartó las mutaciones patógenas en los genes de desmosoma 5, LMNA y MYBPC3. Sin embargo, se identificó una mutación patógena en el gen PLN, la mutación p.Arg14del. No había otros antecedentes familiares relevantes de miocardiopatía o muerte súbita.
El examen de detección sistemática familiar en cascada identificó a otros 7 portadores de la mutación PLN p.Arg14del:
- 1.
Un hermano de 25 años asintomático (figura 1, III.1) tenía extrasístoles ventriculares frecuentes (> 1.000/24 h). El ECG mostró una transición tardía de la onda R en las derivaciones precordiales. Aunque la ecocardiografía era normal, la resonancia magnética cardiaca reveló un vértice ventricular derecho hipocinético y una mancha de contraste de gadolinio subepicárdica tardía en la pared lateral del ventrículo izquierdo. La función sistólica biventricular era normal. Se documentaron episodios recurrentes de taquicardia ventricular no sostenida (15-20 latidos) sintomática. Se inició tratamiento con bloqueadores beta y se implantó un cardioversor-desfibrilador implantable.
- 2.
La madre del probando, de 52 años (II.1), estaba asintomática. El ECG mostró una transición de la onda R tardía en las derivaciones precordiales. Los resultados de la ecocardiografía y la resonancia magnética cardiaca fueron normales.
- 3.
La abuela materna (I.1) fue diagnosticada incidentalmente a la edad de 74 años en una evaluación cardiaca preoperatoria. El ECG mostró ondas T negativas en las derivaciones inferiores y laterales. La ecocardiografía mostró fracción de eyección del ventrículo izquierdo del 45%, con unos diámetros del ventrículo izquierdo de tamaño normal. La ecocardiografía en esfuerzo fue negativa para isquemia, y el registro Holter no mostró arritmia. La paciente presentó buena respuesta al tratamiento médico, con normalización de la fracción de eyección del ventrículo izquierdo.
- 4.
Tres tías maternas asintomáticas (II.2, II.3 y II.4) presentaban mala progresión de la onda R y ondas T planas en todo el ECG. Los resultados de las ecocardiografías y la resonancia magnética cardiaca fueron normales.
- 5.
Un primo de 16 años (III.3) estaba asintomático, y su estudio diagnóstico fue normal.
Dada la morfología de la taquicardia ventricular y las anomalías del ECG y su estado de portadores, 2 pacientes cumplían los criterios de miocardiopatía ventricular derecha arritmógena definitiva, mientras que 3 tenían un diagnóstico limítrofe.
El análisis de haplotipo para marcadores alrededor del PLN en 2 portadores de la mutación de PLN españoles afectados se comparó con la serie holandesa. Es interesante señalar que los pacientes españoles tenían en común 4 de 5 marcadores del haplotipo común holandés, lo que apunta a un antecesor fundador común.
En la familia presentada aquí, la penetración de la enfermedad es de 6/8 (75%). Dado que la PLN p.Arg14del es una mutación patógena, esta familia constituye un claro ejemplo de la variabilidad del fenotipo clínico en los familiares con miocardiopatía arritmogénica ventricular derecha4. Es de destacar que la evidencia reciente muestra una tendencia a que las mujeres portadoras presenten fenotipos más leves que los varones, aunque sufren de todos modos arritmias ventriculares malignas5. Una observación notable fue la gran variedad de anomalías del ECG observadas en la familia: bajo voltaje del QRS generalizado (que recuerda incluso la miocardiopatía restrictiva) en el probando, ondas T invertidas en las derivaciones inferolaterales (típicas de la miocardiopatía ventricular izquierda arritmogénica) y mala progresión de la onda R en todos los portadores excepto uno.
El bajo voltaje y la mala progresión de la onda R en el ECG constituyen alertas para los pacientes con miocardiopatía ventricular derecha arritmogénica o miocardiopatía dilatada y debe sospecharse que la causa de la enfermedad es una mutación del PLN. Esto es de capital importancia, en especial cuando se evalúa el momento apropiado para implantar de un desfibrilador automático implantable que, a la vista de los datos actuales, debe indicarse antes de que el paciente presente una disfunción sistólica grave5.