Palabras clave
INTRODUCCIÓN
En los últimos años, un número creciente de observaciones ha testificado el papel capital que desempeña la inflamación en la patogenia de la aterosclerosis y de sus complicaciones, hasta el punto de que, actualmente, la aterosclerosis debe considerarse una «enfermedad inflamatoria» a todos los efectos1-4. Los datos que se han ido acumulando demuestran que la concentración elevada de marcadores circulantes de inflamación predice una respuesta cardiovascular desfavorable en individuos asintomáticos, en pacientes con cardiopatía isquémica estable y en pacientes con síndromes coronarios agudos5-25. Un mayor conocimiento de los mecanismos moleculares y celulares de la inflamación puede no solamente mejorar la estratificación pronóstica, sino también identificar nuevas dianas terapéuticas4,26. En esta revisión se resumen los mecanismos de la respuesta inflamatoria en los síndromes coronarios agudos, sus implicaciones clínicas y las potenciales estrategias terapéuticas para contrarrestar este fenómeno.
INFLAMACIÓN Y ATEROGÉNESIS
Los desencadenantes de la inflamación en la aterogénesis incluyen los factores de riesgo clásicos, como la hipercolesterolemia27-36, la hipertensión37-42, la diabetes43,44, la obesidad45, la hiperhomocisteinemia46-51, el tabaquismo52-59 y las infecciones60-66. Estos estímulos aterogénicos provocan un daño en la pared vascular y, de acuerdo con la teoría de «respuesta al daño» propuesta por Ross, la aterosclerosis es el resultado de un respuesta exagerada de tipo inflamatorio-fibroproliferativa1,2. La respuesta inflamatoria no sólo promueve el inicio de un proceso aterosclerótico, sino que también contribuye al posterior crecimiento del ateroma y a la precipitación de sucesos trombóticos agudos1-4,67.
El endotelio arterial normal en contacto con la sangre circulante resiste la adhesión firme de leucocitos, incluidos los monocitos sanguíneos68,69. Después de la activación inflamatoria, las células endoteliales aumentan la expresión de varios tipos de moléculas de adhesión leucocitarias, lo que favorece la adhesión y posterior migración de monocitos y linfocitos T a través de las células endoteliales hacia la pared arterial70-72. Varias citocinas quimiotácticas también participan en la movilización de monocitos y linfocitos73-77. Una vez que los monocitos se instalan en la pared arterial, adquieren características de macrófagos tisulares y células espumosas que secretan especies reactivas del oxígeno, citocinas proinflamatorias, metaloproteinasas (MMP), factores de crecimiento y factor tisular, lo que amplifica el proceso inflamatorio local78-81. En la pared arterial, las células T pueden interaccionar con antígenos del tipo de lipoproteínas de baja densidad (LDL) oxidadas y proteínas de estrés térmico (endógenas o microbianas), lo que conduce a la activación de los leucocitos y a la producción de citocinas27,61,82,83. En particular, los linfocitos T coadyuvantes que están dentro del ateroma pueden convertirse en células secretoras de citocinas proinflamatorias (interleucina 1 [IL-1], factor de necrosis tumoral [TNF], interferón gamma [IFN-γ]), conocidas como células TH1, o en células secretoras de citocinas antiinflamatorias (IL-4, IL-10), conocidas como células TH284. Las células TH1 que producen IFN-γ, una citocina pleiotrópica implicada en la activación monocito/macrófago, son las que en general predominan en un ateroma.
En las placas de ateroma maduras se puede, identificar 2 regiones diferentes: la cápsula fibrosa, rica en fibras de colágeno y células musculares lisas, y el núcleo, rico en células espumosas, macrófagos y restos celulares necróticos1-4. Los macrófagos se pueden agrupar en un núcleo central formando una placa de ateroma típica, donde pueden sufrir apoptosis dando lugar al «núcleo necrótico» de la lesión aterosclerótica, o pueden liberar MMP, produciendo la degradación de la matriz extracelular y promoviendo la rotura de la placa85-88. Esto permite que la sangre entre en contacto con el factor tisular (TF), una potente proteína procoagulante que también es producida por los macrófagos, lo que induce la aparición de complicaciones trombóticas67,78-80. Incluso en ausencia de fisuras en la placa, las citocinas proinflamatorias (IL-1, IL-6, TNF-α) pueden potenciar las propiedades procoagulantes de las células endoteliales y los neutrófilos, y contribuir así a las complicaciones trombóticas de la placa de ateroma67,78-83.
En resumen, la aterosclerosis es un proceso crónico con un importante componente inflamatorio activo y en evolución. La inflamación desempeña un papel crítico no sólo como desencadenante del proceso aterosclerótico, sino también promoviendo el desarrollo de la placa de ateroma y de sus complicaciones. Es importante señalar que la evolución de la enfermedad no es uniforme entre los distintos individuos, probablemente debido a diferencias idiosincrásicas en la respuesta inflamatoria a los estímulos aterogénicos. Los individuos con una respuesta inflamatoria mayor a los estímulos aterogénicos tienen un riesgo más elevado de desarrollar manifestaciones clínicas de aterosclerosis. De hecho, los marcadores sistémicos de inflamación, como la proteína C reactiva, están asociados a un riesgo mayor, a largo plazo, de padecer infarto agudo de miocardio, accidente cerebrovascular o enfermedad vascular periférica severa. Mientras que los desencadenantes inflamatorios y los mecanismos de las fases iniciales de aterogénesis son relativamente bien conocidos, los desencadenantes inflamatorios y los mecanismos de las complicaciones trombóticas agudas de las placas de ateroma son probablemente diferentes y todavía se desconocen.
TRANSICIÓN DESDE UN SÍNDROME CORONARIO ESTABLE A UNO INESTABLE: OBSERVACIONES CLÍNICAS Y POST MORTEM
No está claro por qué muchos pacientes con una aterosclerosis severa y extensa permanecen estables durante años sin llegar a desarrollar un síndrome coronario agudo, mientras que otros desarrollan un cuadro agudo como una de las primeras manifestaciones de cardiopatía isquémica a pesar de tener una aterosclerosis coronaria menos severa89,90. Desde el punto de vista clínico, los síndromes coronarios agudos se caracterizan por un comienzo súbito, y por una posible recurrencia de los episodios isquémicos durante un período que puede durar días, semanas o meses, para regresar luego a una fase estable o quiescente de cardiopatía isquémica91,92. Así pues, la presentación clínica indica que los síndromes coronarios agudos están relacionados con altibajos en los estímulos desestabilizadores. La presentación clínica y la evolución, no obstante, pueden variar considerablemente; esta variabilidad puede ser el reflejo de una diferente prevalencia de los mecanismos subyacentes de inestabilidad91,92. En un extremo se encuentran los pacientes con un infarto agudo de miocardio sin previo aviso, que luego permanecen asintomáticos durante años. En el otro extremo están los pacientes en los que el infarto agudo de miocardio va precedido por un cuadro de angina inestable que dura días o semanas, y que posteriormente continúa en forma de episodios recurrentes de inestabilidad y/o reinfarto durante semanas o meses a pesar de recibir los tratamientos más avanzados89,91,92.
Conviene señalar que en una proporción no despreciable de pacientes, los síndromes coronarios agudos se asocian a una reactivación inflamatoria detectable a partir de determinaciones de marcadores sistémicos de inflamación, como la proteína C reactiva. La prevalencia de una concentración elevada de proteína C reactiva (> 3 mg/l) en la sangre periférica oscila entre un 70% en pacientes con angina inestable severa y cerca de un 100% en los casos de infarto agudo de miocardio precedido por angina inestable, mientras que se encuentra en menos del 50% en los casos de infarto agudo de miocardio no precedido por angina inestable, y en menos de un 20% en pacientes con angina estable17,18,89,93.
Los estímulos desestabilizadores, independientemente de su naturaleza, causan una trombosis coronaria oclusiva que es la responsable directa de la isquemia miocárdica (fig. 1) 94. El hallazgo post mortem más llamativo y característico de la angina inestable comparado con la angina estable crónica es la frecuente presencia de trombos coronarios murales no oclusivos en el lugar de la rotura de las placas de ateroma, que se acompaña ocasionalmente de embolización distal95-97. Los trombos murales están compuestos por plaquetas y fibrina, y a menudo crecen a partir de la pérdida de continuidad endotelial en una placa fisurada. Es importante indicar que: a) los trombos se componen con frecuencia de capas de diferentes edades, lo que es indicativo de un fenómeno trombótico que se desarrolla en distintas etapas en intervalos de días o semanas; b) en un 25-50% de los casos no hay fisura de la placa, sino solamente erosión endotelial que puede identificarse bajo el trombo, y c) en ocasiones no se puede encontrar el trombo95,97. Además, se pueden encontrar pequeñas fisuras de la placa con trombos plaquetarios intraintimales en alrededor del 10% de los sujetos que fallecen por causas no cardíacas y en aproximadamente el 20% de los individuos con hipercolesterolemia, hipertensión y diabetes95,98,99. A diferencia de la angina estable, otros hallazgos distintivos que se han observado en la lesión causante en la angina inestable son: a) un aumento de la concentración de células inflamatorias, incluidos los linfocitos T activados, macrófagos y mastocitos; b) un aumento de la hiperplasia celular; c) un aumento de la inmunorreactividad de la endotelina 1, y d) bandas de contracción en las células musculares lisas circundantes98,100-109.

Fig. 1. Papel de la inflamación en la aterogénesis y en la transición desde los síndromes coronarios estables a los inestables.
El espasmo coronario debido a la hiperreactividad de las células musculares lisas es la causa predominante de infarto de miocardio en pacientes con una historia de angina vasospástica, aunque este suceso es raro110,111. Sin embargo, la vasoconstricción coronaria y la trombosis están profundamente interrelacionadas. Por una parte, se sabe que el espasmo coronario oclusivo y el estancamiento sanguíneo distal causan un incremento significativo y transitorio del fibrinopéptido A en la sangre sistémica112,113. Por otra parte, se sabe que la serotonina, una sustancia liberada por las plaquetas activadas, produce espasmo oclusivo en pacientes con angina inestable e isquemia debido a una constricción vascular distal114. Este círculo vicioso, probablemente mediado por la serotonina, el tromboxano A2, la trombina y la endotelina, puede desempeñar un papel importante en el establecimiento de la angina inestable, donde las placas coronarias inestables, que muestran a menudo unas células musculares lisas preservadas, entran en contacto con las plaquetas activadas. Además, diversos hallazgos dan soporte a la posible hiperreactividad de las células musculares lisas en la angina inestable. De hecho, las placas inestables parecen ser más reactivas a los estímulos del ejercicio y a la prueba del frío que las placas estables, particularmente cuando hay una elevación de los marcadores sistémicos de inflamación115-119.
INFLAMACIÓN EN LOS SÍNDROMES CORONARIOS AGUDOS
Evidencia de inflamación
Pocos años atrás, la observación ocasional de unas estrías rojas a lo largo de las principales vías coronarias durante la cirugía de bypass en pacientes inestables106 y la observación de células inflamatorias infiltradas en el lugar de las placas y en los nervios perivasculares105,106 apuntaron a la intrigante posibilidad de que la inflamación contribuyese a los síndromes coronarios estimulando o aumentando las respuestas hemostáticas y vasoconstrictoras locales.
Estas observaciones post mortem fueron posteriormente confirmadas por una serie de estudios clínicos, que mostraron una activación sistémica de las células inflamatorias. Dinerman et al120 encontraron un aumento sistémico de la concentración sanguínea de la elastasa de los neutrófilos, y Biasucci et al121 hallaron una reducción de la peroxidación intracelular de los neutrófilos, ambos parámetros indicativos de activación de neutrófilos, en pacientes con angina inestable o infarto agudo de miocardio en comparación con los pacientes que tenían angina estable crónica. Mazzone et al encontraron un aumento de la expresión de la molécula de adhesión CD11b/18 en la superficie de monocitos y granulocitos obtenidos en el seno coronario de pacientes con angina inestable comparado con la observada en pacientes con angina estable122; por el contrario, la expresión de CD11b/18 en las muestras aórticas fue similar en los 2 grupos de pacientes, lo que sugiere una activación transcardíaca de los monocitos y granulocitos en la angina inestable. Serneri et al123 demostraron que los monocitos humanos cocultivados con linfocitos obtenidos de pacientes con angina inestable exhibían una actividad procoagulante mayor que la observada en los monocitos cocultivados con linfocitos obtenidos de pacientes con angina estable o pacientes del grupo control, lo que sugiere una activación linfocitaria en la angina inestable.
Berk et al124 describieron un aumento de la concentración sanguínea de proteína C reactiva en los pacientes con angina inestable comparados con los que tenían angina estable. Conviene recordar que la proteína C reactiva es el reactante prototípico de la fase aguda y es sintetizada por el hígado después de la estimulación con IL-6, que es producida fundamentalmente por los monocitos activados; la concentración sanguínea de proteína C reactiva empieza a aumentar cerca de 6 h después de la estimulación hepática125. Y lo que es más importante, Liuzzo et al hallaron un aumento de la concentración sanguínea de proteína C reactiva en pacientes con infarto agudo de miocardio hospitalizados durante las primeras 6 h desde el inicio de los síntomas, y en pacientes con angina inestable y baja concentración de troponina, en los que el incremento de proteína C reactiva no puede deberse a una necrosis miocárdica17. Este importante estudio sugiere fuertemente y por primera vez que la activación súbita de las células inflamatorias puede desempeñar un papel crítico en la patogenia de los síndromes coronarios agudos.
Datos procedentes de nuestro grupo han confirmado que la reactivación inflamatoria asociada a los síndromes coronarios agudos no es un epifenómeno, sino más bien un componente patogénico fundamental. De hecho, no es solamente que la inflamación no se pueda atribuir a la necrosis celular miocárdica17, sino que tampoco se relaciona con la severidad de la aterosclerosis, puesto que no hay correlación entre el grado de aterosclerosis y la respuesta en la fase aguda en pacientes con angina estable crónica o enfermedad vascular periférica126. La inflamación no puede atribuirse a la activación episódica del sistema hemostático, ya que la elevación sistémica de los marcadores de la producción de trombina (complejo trombina-antitrombina y fragmento de protrombina 1 + 2) no se acompaña de una elevación posterior de las proteínas de la fase aguda127; tampoco puede ser atribuida al daño por isquemia/reperfusión, puesto que los neutrófilos circulantes no están activados y la concentración de proteína C reactiva no está aumentada en los pacientes con angina variante, a pesar de tener un número significativamente mayor de episodios isquémicos128. Por último, es importante indicar que la inflamación no está relacionada con la rotura de la placa, ya que no se observa un aumento de la IL-6 o de la proteína C reactiva en pacientes estables con valores basales de proteína C reactiva bajos que se someten a una angioplastia, una causa iatrogénica de rotura de la placa129.
Consecuencias de la inflamación de la placa
Con independencia de cuál sea su causa, la reactivación inflamatoria asociada a los síndromes coronarios agudos es la expresión de las células inflamatorias activadas, algunas de las cuales se localizan probablemente en la placa de ateroma causante, desde donde pueden provocar diversos efectos perjudiciales a través de una gran variedad de mecanismos diferentes.
Activación endotelial
Las citocinas liberadas por las células inflamatorias activadas tienen el potencial de activar el endotelio transformando sus propiedades antiadhesivas y anticoagulantes en propiedades adhesivas y procoagulantes1-4. De hecho, las células endoteliales estimuladas por IL-1, TNF o endotoxinas expresan moléculas de adhesión en su superficie, como la molécula de adhesión intercelular 1 (ICAM-1) y la E-selectina, y segregan sustancias quimiotácticas solubles, como la proteína quimiotáctica 1 del monocito (MCP-1), el factor estimulante de las colonias de monocitos (M-CSF) y la IL-81-4. Además, en las células endoteliales activadas, diferentes moléculas de adhesión y quimiotácticas se expresan casi de manera simultánea, lo que sugiere una activación concertada de diferentes genes probablemente relacionados, al menos en parte, con la activación del factor nuclear κB (NF-κB)130. Este factor fue descrito inicialmente en linfocitos, donde controla la activación de genes que codifican las cadenas κ de las inmunoglobulinas. Consiste en una familia de factores de transcripción diméricos ligados a una proteína inhibidora (I-κB). La fosforilación de I-κB produce la translocación de las subunidades activas al núcleo, donde se ligan a secuencias específicas en regiones promotoras de diferentes genes y activan la transcripción de ARNm130-132. Se han encontrado secuencias capaces de unirse a elementos del NF-κB en varios genes humanos, incluidos los que codifican moléculas de adhesión endoteliales. Así, el sistema de NF-κB puede mediar la síntesis endotelial inducida por las citocinas de las moléculas de adhesión y de las sustancias quimiotácticas solubles que se producen después de la activación endotelial130-132.
El endotelio vascular contribuye activamente al equilibrio dinámico entre los estímulos antitrombóticos y protrombóticos68,69. En condiciones normales, las células endoteliales actúan para prevenir la coagulación, pero la incubación con citocinas, como la IL-1 o el TNF-α, produce un aumento de la actividad procoagulante que alcanza un pico a las 4 h y vuelve a su estado basal dentro de las 24 h, probablemente mediado por la expresión de factor tisular133. Los efectos procoagulantes de la IL-1 y el TNF-α parecen ser aditivos, ya que la incubación con las 2 citocinas da lugar a un desarrollo mayor de la actividad procoagulante que la incubación co n cualquiera de los mediadores de forma individual, incluso utilizando las dosis máximas133. Hay que tener en cuenta que la IL-6 aumenta la reactividad plaquetaria que, a su vez, puede potenciar los efectos procoagulantes debidos a la activación endotelial mediada por la IL-1 y el TNF134.
Alteraciones del metabolismo de la matriz extracelular
El principal componente de la cápsula fibrosa de las placas de ateroma es una matriz extracelular densa y fibrosa (antes llamada tejido conectivo)135,136. Los constituyentes fundamentales de esta matriz extracelular son el colágeno de tipo I y III (una espiral helicoidal triple derivada de precursores de procolágeno específicos), la elastina y los proteoglicanos. El IFN-γ elaborado por las células T reduce la síntesis de colágeno, lo que provoca la apoptosis de las células musculares lisas e inhibe específicamente la síntesis de colágeno en las células musculares lisas135,136. Además, los macrófagos cargados de lípidos y estimulados por una gran variedad de citocinas, como el INF-γ, M-CSF, MCP-1 y IL-1, liberan MMP de matriz, como la colagenasa y la estromalisina, favoreciendo la degradación de la matriz intercelular. La colagenasa, la estromalisina y la gelatinasa B se expresan también en otros tipos de células que se encuentran en la placa de ateroma, como las células endoteliales y las células musculares lisas, después de ser activadas por citocinas135,136. Por último, las citocinas no parecen afectar a la síntesis de inhibidores tisulares de las MMP de matriz137.
En resumen, la inflamación de la placa tiene el potencial de favorecer la aparición de fisuras al reducir la concentración de proteínas contenidas en la matriz extracelular.
Hiperreactividad de las células musculares lisas
Zeiher et al138 han demostrado la existencia de una mayor inmunorreactividad de la endotelina-1 en placas coronarias inestables, en comparación con la que se ha encontrado en placas estables obtenidas por aterectomía direccional138. Esta observación puede proporcionar la clave para entender los mecanismos de la hiperreactividad coronaria segmentaria observada a menudo en pacientes con angina inestable, en particular en presencia de una concentración elevada de proteína C reactiva119. De hecho, la endotelina 1 no es solamente una sustancia vasoconstrictora potente, sino que también potencia los efectos de otros estímulos vasoconstrictores, como las catecolaminas, la serotonina y la angiotensina II139. Curiosamente, la endotelina no es sintetizada sólo por las células endoteliales, sino también por los macrófagos humanos y los leucocitos polimorfonucleares estimulados por lipopolisacáridos. Otras evidencias que apoyan la hipótesis de que las células inflamatorias activadas pueden causar hiperreactividad de las células musculares lisas proceden de la observación, en un modelo porcino, de que la envoltura de un segmento coronario proximal con una malla de algodón que contenga bolas de sefarosa absorbidas con IL-1β humana recombinante determina la hiperreactividad local de las células musculares lisas a la serotonina y la histamina en pocas semanas140. Estos cambios funcionales se previenen por un tratamiento simultáneo con anticuerpos neutralizantes contra la IL-1β o el factor de crecimiento derivado de las plaquetas (PDGF), lo que sugiere que el PDGF puede desempeñar un papel importante como mediador de la respuesta vasospástica inducida por IL-1 beta.
Causas de la inflamación
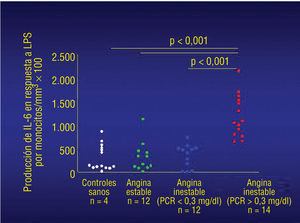
Después de una angioplastia coronaria, o tras el débil estímulo inflamatorio que se produce en la angiografía coronaria, se observa un aumento de IL-6 y de proteína C reactiva en pacientes con angina inestable que tienen concentraciones basales de proteína C reactiva elevadas129. De acuerdo con esto, los monocitos periféricos de los pacientes inestables con concentraciones de proteína C reactiva elevadas (> 0,3 mg/dl) presentan una respuesta in vitro exagerada a la estimulación con lipopolisacáridos, en comparación con la que presentan los monocitos de pacientes inestables con concentraciones bajas de proteína C reactiva (< 0,3 mg/dl), o con la que presentan los pacientes con angina estable o los controles sanos (fig. 2)141. Además, se ha descrito que, en pacientes con infarto agudo de miocardio, la fase aguda de la respuesta proteínica a la necrosis es independiente del tamaño del infarto, pero se puede predecir a partir de la concentración basal de proteína C reactiva; en este estudio se documentó un aumento de la concentración basal de proteína C reactiva en el 85% de los casos de infarto agudo de miocardio precedido por angina inestable93. Considerados en su conjunto, estos hallazgos sugieren que la hiperreactividad de las células inflamatorias a los estímulos inflamatorios subliminales puede contribuir a la inestabilidad coronaria. De acuerdo con esta hipótesis, se ha identificado un subgrupo inusual de células T que expresa el fenotipo nulo CD4+CD28 en pacientes con un estado inflamatorio elevado142. Estas células T inusuales están dirigidas a la producción de IFN-δ.
La regulación crónica aumentada del IFN-γ en pacientes con angina inestable puede conducir a la activación subsiguiente de monocitos/macrófagos en la circulación, así como en las lesiones tisulares delta. El hallazgo de que las células T nulas para CD28 tienen capacidad citolítica sugiere que las reacciones inmunológicas en sujetos con este tipo de células T tienen un riesgo elevado de producir daño tisular. Tanto factores ambientales como mecanismos genéticos pueden subyacer a las perturbaciones del repertorio de células T. Puesto que los defectos en la expresión de la membrana celular de CD28 pueden aparecer como consecuencia de una exposición crónica al antígeno, la expansión de células T nulas para CD4+CD28 puede ser reflejo de una respuesta inmunológica persistente a los microorganismos o a autoantígenos contenidos en las placas de ateroma142,143.

Fig. 2. Producción de interleucina-6 (IL-6) en monocitos después de la estimulación in vitro con lipopolisacárido (LPS) en pacientes con angina inestable que tienen una concentración elevada (> 0,3 mg/dl) de proteína C reactiva (PCR), en pacientes con angina inestable que tienen una concentración baja (< 0,3 mg/dl) de proteína C reactiva, en pacientes con angina estable y en controles sanos.
Es importante enfatizar que la inflamación asociada a los síndromes coronarios agudos es un fenómeno general que no está restringido a la lesión causante. De acuerdo con este concepto, estudios previos han demostrado que los síndromes coronarios agudos se asocian a trombosis coronarias múltiples en el examen post mortem, con deterioro microvascular en regiones remotas y con un aumento de la progresión a corto plazo de estenosis no causantes95. Además, Goldstein et al144 han encontrado que dos quintas partes de los pacientes con infarto agudo de miocardio presentan múltiples placas coronarias complejas, que se asocian a consecuencias clínicas adversas. En un estudio post mortem más reciente, de Spagnoli et al145 utilizaron una técnica novedosa para la determinación cuantitativa de componentes celulares de las arterias coronarias epicárdicas y demostraron la existencia de una activación difusa de células inflamatorias, tanto en las arterias relacionadas con el infarto como en las no relacionadas, en pacientes con infarto agudo de miocardio, pero no en pacientes que tenían un infarto antiguo145. Por último, Buffon et al146 describieron una activación generalizada de neutrófilos a lo largo de todo el lecho vascular en pacientes con angina inestable, con independencia de la localización de la estenosis causante146.
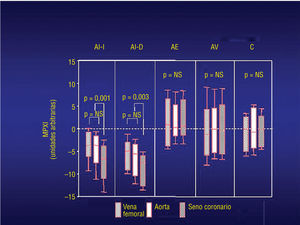
En concreto, el contenido de mieloperoxidasa procedente de neutrófilos en muestras sanguíneas obtenidas de la gran vena cardíaca, que drena sangre de forma selectiva desde la arteria coronaria izquierda pero no desde la derecha, estaba significativamente disminuido en pacientes con angina inestable, con independencia del lugar de la estenosis (arteria coronaria izquierda o derecha), pero no lo estaba en pacientes con angina estable, estenosis múltiples, angina variante e isquemia recurrente, o en controles sanos (fig. 3). Es más, estas observaciones patológicas, angiográficas y clínicas ponen en duda seriamente el concepto de una placa vulnerable única en los síndromes coronarios inestables, y dan soporte a la idea de que la inestabilidad de las placas no es meramente un accidente vascular local, sino que es probable que refleje un proceso fisiopatológico más generalizado, que tiene el potencial de desestabilizar placas de ateroma a lo largo del árbol coronario.

Fig. 3. Activación de neutrófilos, expresada como cambios en el contenido de mieloperoxidasa (MPXI), obtenidos a partir de la gran vena cardíaca (seno coronario), aorta y arteria femoral en pacientes con angina inestable (AI) con enfermedad arterial coronaria izquierda (AI-I) y derecha (AI-D), en pacientes con angina estable (AE), en pacientes con angina variante (AV) y en controles (C).
Todavía no se ha podido identificar los desencadenantes de la generalización de la inflamación asociada a los síndromes coronarios agudos. Caligiuri et al147 han demostrado que el repertorio de receptores de antígenos de las células T activadas estaba sesgado en el 57% de los pacientes con angina inestable, frente al 23% de los pacientes con cardiopatía isquémica estable, lo que apoya la hipótesis de que una respuesta inmunológica provocada por un antígeno puede desempeñar un papel crítico en la patogenia de la inestabilidad coronaria147. La observación reciente, por parte de Biasucci et al148, de la seropositividad a la proteína 60 de choque térmico de Chlamydia pneumoniae en el 98% de los pacientes con angina estable y en el 0% de los controles sugiere que la infección por C. pneumoniae que se acompaña de una expresión de la proteína 60 de choque térmico puede ser un potencial desencadenante, posiblemente a través de un mimetismo antigénico (fig. 4)148.

Correspondencia: Prof. F. Crea.
Institute of Cardiology-Catholic University of the Sacred Heart.
Largo Agostino Gemelli, 8. 00168 Rome. Italy.
Correo electrónico: fcrea@rm.unicatt.it