El gen PLAU, que codifica para el activador del plasminógeno tipo urocinasa, desempeña un papel destacado en el crecimiento colateral. Se ha investigado si el polimorfismo PLAU P141L (C>T), que causa una mutación en el dominio kringle de la proteína, se asocia con la circulación colateral coronaria en una cohorte de 676 pacientes con enfermedad arterial coronaria.
MétodosSe genotipificó el polimorfismo de muestras de sangre mediante prueba basada en TaqMan, y la circulación colateral se evaluó por el método Rentrop. Las asociaciones de las variantes alélicas y los genotipos con la circulación colateral se examinaron mediante modelos de regresión logística multivariable ajustados por las variables clínicamente relevantes.
ResultadosLos pacientes con circulación colateral deficiente (Rentrop 0-1; n = 547) presentaron mayor frecuencia del genotipo TT que aquellos con buena circulación colateral (Rentrop 2-3; n = 129; p = 0,020). Por otra parte, el alelo T fue más frecuente en los paciente con circulación deficiente (p=0,006). La odds ratio de los portadores del alelo T de presentar una circulación colateral deficiente (ajustada por variables clínicamente relevantes) fue estadísticamente significativa en el modelo dominante (odds ratio=1,83 [intervalo de confianza del 95%, 1,16-2,90]; p = 0,010) o el aditivo (odds ratio=1,73 [intervalo de confianza del 95%, 1,14-2,62]; p = 0,009).
ConclusionesSe demuestra una asociación entre la circulación colateral coronaria y el polimorfismo PLAU P141L. Los pacientes con la variante 141L tienen mayor riesgo de tener una circulación colateral deficiente.
Palabras clave
La enfermedad arterial coronaria (EAC) es la principal causa de muerte en los países industrializados1–4. En la EAC, el crecimiento de las arterias colaterales coronarias (arteriogénesis coronaria) se ha reconocido como una fuente alternativa de aporte sanguíneo a un área miocárdica afectada por la isquemia1,3. Los pacientes con niveles elevados de colateralización tienen un 36% menos riesgo de mortalidad que los pacientes con bajos niveles de colateralización5. Un desencadenante importante en la etiología de la formación de arterias colaterales son la isquemia crónica y la extensión de la carga aterosclerótica. Aun así, la capacidad de los pacientes con EAC para inducir el crecimiento colateral es muy heterogéneo, incluso en aquellos con arterias totalmente ocluidas6, y probablemente se trata de un proceso determinado genéticamente7,8.
Fulton9 demostró que el corazón humano contiene una gran red colateral preexistente incluso antes de la aparición de la enfermedad aterosclerótica obstructiva (revisado por Van Royen et al10). La arteriogénesis se basa en la presencia de estas arteriolas colaterales que interconectan ramas laterales proximal y distalmente a la arteria ocluida (anastomosis)10,11. Tras la oclusión de una arteria, la redirección del flujo sanguíneo a través de conexiones arteriolares preexistentes desencadena, en primera instancia, el aumento de fuerzas mecánicas tales como la tensión de cizallamiento y la tensión circunferencial de la pared, a través del gradiente de presión formado entre la región de alta presión, proximal a la oclusión, y la región de baja presión, distal11–13. Estas fuerzas inducen el crecimiento colateral coronario. Este proceso supone la remodelación estructural completa de la pared arterial, que a su vez comprende la proliferación de las células endoteliales y de las células de la musculatura lisa y, finalmente, la reorganización del componente extracelular y de la lámina elástica interna. Juntos, estos procesos conducen a una reducción en la tensión de cizallamiento de la pared arterial hasta valores basales fisiológicos14.
Las células endoteliales perciben la tensión de cizallamiento y transforman esta señal en cambios en la expresión génica11–15. De este modo, el endotelio se activa, y se promueve la atracción y la adhesión de los monocitos que secretarán factores de crecimiento y citocinas a las arterias en crecimiento11. En su fase temprana, el crecimiento arterial colateral se asocia con un aumento de la expresión del receptor del factor de crecimiento de fibroblastos-1 en el sitio del vaso donde tiene lugar la remodelación, mientras que los monocitos suministran por vía paracrina los ligandos (factores de crecimiento de fibroblastos) que promueven el crecimiento15. La activación del receptor del factor de crecimiento de fibroblastos-1 por factores de crecimiento de fibroblastos por factores de crecimiento de fibroblastos origina la activación de la vía de Ras/Raf, MEK1/2, ERK1/2 y, finalmente, el aumento de la expresión del factor de respuesta de crecimiento temprano-1, implicado en la iniciación de un fenotipo migratorio e invasivo en las células endoteliales y en las del músculo liso. El factor de respuesta de crecimiento temprano-1 induce la expresión del activador del plasminógeno tipo urocinasa (uPA)14.
El uPA es fundamental en la regulación de la adhesión celular, la migración y la proliferación16 y la remodelación de la pared arterial debida a lesiones mecánicas y durante la arteriogénesis16–19. El uPA convierte el plasminógeno en plasmina, que a su vez activa factores de crecimiento, formas latentes de citocinas y metaloproteinasas de matriz 2 y 9, que participan en la degradación, estrictamente localizada, de la matriz extracelular, crucial para permitir la migración de células musculares lisas y la formación de la neoíntima16,17 durante el crecimiento colateral.
PLAU, el gen que codifica para uPA, se localiza en el cromosoma 10q22.2 entre dos regiones que muestran vinculación con la enfermedad de Alzheimer20. El polimorfismo de un solo nucleótido (SNP) rs2227564 (un polimorfismo C/T de la segunda base del codón 141 del gen PLAU) causa una mutación de cambio de sentido (de prolina a leucina) en el dominio kringle de uPA, que tiene un efecto funcional en la unión del cimógeno de la fibrina a uPA21. Aunque este SNP se ha asociado con varias enfermedades22–29, su asociación con la arteriogénesis coronaria no se ha estudiado hasta la fecha. El objetivo de nuestro estudio es evaluar la asociación del polimorfismo PLAU P141L con el crecimiento arterial colateral coronario, planteando la hipótesis que la variante PLAU L141 se asocia con una respuesta arteriogénica reducida en los pacientes con EAC.
MÉTODOSEstudio y selección de los pacientesEl estudio se realizó de conformidad con la Declaración de Helsinki y el protocolo aprobado por el Comité de Bioética del Centre Cardiovascular Sant Jordi y el Hospital Universitari de la Vall d’Hebron para el periodo comprendido entre 2008 y 2012. Todos los pacientes dieron su consentimiento informado y se obtuvo su autorización por escrito.
Se seleccionó a los participantes en el estudio consecutivamente de entre los pacientes sometidos a cateterización diagnóstica de las arterias coronarias. Se incluyó una cohorte de 677 pacientes con EAC y estenosis grave (≥ 70%). Se excluyó a los pacientes con infarto agudo de miocardio reciente (< 1 mes), anemia, angioplastia reciente, revascularización previa por intervención coronaria percutánea, cirugía de bypass de la arteria coronaria, infección o inflamación o insuficiencia renal crónica. Se estudió con detalle la historia clínica de cada paciente y se registraron las características demográficas, clínicas y de laboratorio de cada uno. Hipertensión, diabetes mellitus (DM), tipo de DM, hipercolesterolemia e hipertrigliceridemia, antecedentes de tabaquismo, antecedentes familiares de cardiopatías, antecedentes de angina de pecho y tipo de angina e infarto agudo de miocardio se registraron como variables categóricas (presencia o ausencia).
Angiografía coronaria y evaluación de la circulación colateral coronariaLa angiografía coronaria se realizó en múltiples proyecciones ortogonales utilizando la técnica de Judkins. La circulación colateral coronaria (CCC) se evaluó angiográficamente utilizando el método de Rentrop et al30. Para evaluar el llenado de las colaterales, se utilizó la siguiente escala: 0, sin llenado visible de colaterales; 1, llenado colateral de ramas del vaso que se ha de dilatar sin que el contraste alcance el segmento epicárdico de este vaso; 2, llenado parcial colateral del segmento epicárdico del vaso que se está dilatando, y 3, llenado completo del segmento epicárdico del vaso que se está dilatando.
Se clasificó a los pacientes con EAC según el grado de CCC: deficiente (Rentrop 0-1; n=546) y bien desarrollada (Rentrop 2-3; n=131). La CCC fue evaluada por tres cardiólogos hemodinamistas experimentados que no recibieron información previa sobre los pacientes. El grado de concordancia en la evaluación diagnóstica de la CCC entre los tres evaluadores fue alto y se determinó a través del estadístico kappa (κ=0,987; intervalo de confianza del 95% [IC95%], 0,953-1,000; p<0,001). Este test estadístico se determinó con las primeras 100 angiografías revisadas.
Análisis genotípicoSe extrajo sangre a los pacientes en el momento de la coronariografía. El ADN genómico se aisló utilizando la estación de trabajo automatizada Chemagen (Baesweiler, Alemania), siguiendo el protocolo del fabricante. Para determinar el genotipo del polimorfismo rs2227564 en las muestras de sangre, se realizó un análisis TaqMan de genotipificación de SNP (Applied Biosystems; Foster City, California, Estados Unidos) utilizando el sistema de reacción en cadena de la polimerasa en tiempo real Fast 7900HT (Applied Biosystems). Las determinaciones de los genotipos de cada paciente se reprodujeron en tres pruebas independientes.
Análisis estadísticoLos datos continuos de distribución no normal se analizaron mediante la prueba de la U de Mann-Whitney. En este estudio, la edad no mostró una distribución normal (Shapiro-Wilk, p<0,001). Las asociaciones entre datos categóricos se evaluaron utilizando la prueba exacta de Fisher o la de la χ2, y el equilibrio de Hardy-Weinberg se evaluó mediante la prueba de la χ2. Para estimar las odds ratio (OR) y sus IC95% entre los genotipos y el riesgo de una CCC reducida, se ajustaron modelos de regresión logística multivariable utilizando las variables clínicamente relevantes. Con el modelo de regresión multivariable, también se analizaron los términos de interacción entre el SNP y las covariables significativas. Los análisis estadísticos se realizaron utilizando el software STATA 11.2. Se estimó la potencia para detectar una asociación genética utilizando el mismo paquete estadístico.
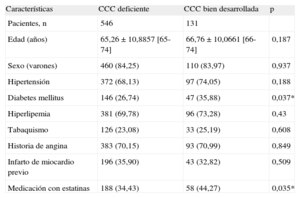
RESULTADOSEl análisis estadístico mostró que no hay diferencias de distribución estadísticamente significativas entre los grupos con CCC deficiente y CCC bien desarrollada en cuanto a edad, sexo, hipertensión o antecedentes de hiperlipemia, tabaquismo, angina o infarto de miocardio previo. La prevalencia de DM (35,88%) y el porcentaje de pacientes con prescripción con estatinas (44,27%) fueron significativamente mayores en el grupo de CCC reducida (tabla 1). En nuestra población, 462 pacientes (68,24%) presentaban el genotipo CC, 194 (28,66%) eran heterocigotos (CT) y 21 (3,01%) tenían el genotipo TT. Las frecuencias de los alelos C y T en el total de la muestra fueron del 82,57 y el 17,43% respectivamente (tabla 2). Estas frecuencias son compatibles con el equilibrio de Hardy-Weinberg (χ2=0,059; p=0,8077).
Características clínicas de los pacientes con enfermedad arterial coronaria en función de la circulación colateral coronaria
| Características | CCC deficiente | CCC bien desarrollada | p |
| Pacientes, n | 546 | 131 | |
| Edad (años) | 65,26±10,8857 [65-74] | 66,76±10,0661 [66-74] | 0,187 |
| Sexo (varones) | 460 (84,25) | 110 (83,97) | 0,937 |
| Hipertensión | 372 (68,13) | 97 (74,05) | 0,188 |
| Diabetes mellitus | 146 (26,74) | 47 (35,88) | 0,037* |
| Hiperlipemia | 381 (69,78) | 96 (73,28) | 0,43 |
| Tabaquismo | 126 (23,08) | 33 (25,19) | 0,608 |
| Historia de angina | 383 (70,15) | 93 (70,99) | 0,849 |
| Infarto de miocardio previo | 196 (35,90) | 43 (32,82) | 0,509 |
| Medicación con estatinas | 188 (34,43) | 58 (44,27) | 0,035* |
CCC: circulación colateral coronaria.
Los datos se expresan como n (%), media±desviación estándar o mediana [intervalo intercuartílico].
*Se consideró estadísticamente significativo un valor p<0,05.
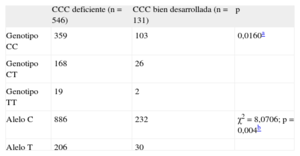
Asociación de las distribuciones genotípicas y alélicas del polimorfismo PLAU P141L (rs2227564) en pacientes con enfermedad arterial coronaria en función de la circulación colateral coronaria
| CCC deficiente (n=546) | CCC bien desarrollada (n=131) | p | |
| Genotipo CC | 359 | 103 | 0,0160a |
| Genotipo CT | 168 | 26 | |
| Genotipo TT | 19 | 2 | |
| Alelo C | 886 | 232 | χ2=8,0706; p=0,004b |
| Alelo T | 206 | 30 |
CCC: circulación colateral coronaria.
Un valor p<0,05 se consideró estadísticamente significativo.
El análisis del polimorfismo PLAU P141L (C>T) mostró asociación entre la distribución del genotipo y el grado de CCC (prueba exacta de Fisher, p=0,0160). La frecuencia del genotipo TT entre los pacientes con EAC y CCC deficiente fue mayor que entre los que mostraron una CCC bien desarrollada. Por otra parte, la variante alélica T también fue más frecuente en pacientes con CCC deficiente (χ2=8,0706; p=0,004) (tabla 2). Esta asociación se mantuvo también en el subgrupo de pacientes con estenosis de alto grado (≥ 95%) (p=0,016).
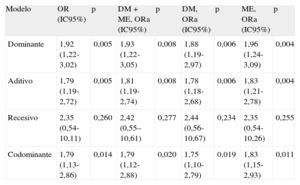
Las OR de tener CCC deficiente para los pacientes portadores del alelo T que codifica para la forma L141 en el modelo dominante (CT+TT frente a CC) (OR=1,92; IC95%, 1,22-3,02; p=0,005) y en el modelo aditivo (OR=1,79; IC95%, 1,19-2,72; p=0,005) mostraron significación estadística (tabla 3). Cuando se ajustó por posibles variables de confusión como la DM y la medicación con estatinas (tabla 1), las OR de tener CCC deficiente para los pacientes portadores el alelo T en el modelo dominante (CT+TT frente a CC) (OR=1,93; IC95%, 1,22-3,05; p=0,008) y el modelo aditivo (OR=1,81; IC95%, 1,19-2,74; p=0,008) también fueron estadísticamente significativas (tabla 3). Cuando los datos se ajustaron por separado por DM o por medicación con estatinas, se obtuvieron resultados similares (tabla 3). Considerando que la diferencia del porcentaje de desviación entre la OR bruta y la OR ajustada por DM y/o medicación con estatinas estaba por debajo de 10%, se concluye que estas variables no son factores de confusión.
Odds ratio del polimorfismo de un solo nucleótido PLAU P141L (rs2227564) en pacientes con enfermedad arterial coronaria con respecto a la circulación colateral coronaria según diferentes modelos de herencia
| Modelo | OR (IC95%) | p | DM+ME, ORa (IC95%) | p | DM, ORa (IC95%) | p | ME, ORa (IC95%) | p |
| Dominante | 1,92 (1,22-3,02) | 0,005 | 1,93 (1,22-3,05) | 0,008 | 1,88 (1,19-2,97) | 0,006 | 1,96 (1,24-3,09) | 0,004 |
| Aditivo | 1,79 (1,19-2,72) | 0,005 | 1,81 (1,19-2,74) | 0,008 | 1,78 (1,18-2,68) | 0,006 | 1,83 (1,21-2,78) | 0,004 |
| Recesivo | 2,35 (0,54-10,11) | 0,260 | 2,42 (0,55–10,61) | 0,277 | 2,44 (0,56-10,67) | 0,234 | 2,35 (0,54-10,26) | 0,255 |
| Codominante | 1,79 (1,13-2,86) | 0,014 | 1,79 (1,12-2,88) | 0,020 | 1,75 (1,10-2,79) | 0,019 | 1,83 (1,15-2,93) | 0,011 |
DM: diabetes mellitus; IC95%: intervalo de confianza del 95%; ME: medicación con estatinas; OR: odds ratio; ORa: odds ratio ajustada.
Un valor p<0,05 se consideró estadísticamente significativo.
El análisis de la interacción entre el SNP y covariables significativas no reveló asociación alguna. Asumiendo las tasas del 18,86 y el 11,45% de presencia del alelo T en pacientes con CCC deficiente y bien desarrollada respectivamente, la potencia estadística para detectar diferencias genéticas fue del 81,6%.
DISCUSIÓNLa comprensión del programa genético que conduce al desarrollo de las arterias colaterales es de una gran importancia para mejorar el diagnóstico, el tratamiento y la prevención de la EAC7,8. Las colaterales coronarias pueden ser un marcador pronóstico útil: los pacientes con escasa colateralización tienen mayor riesgo de muerte y se debe seguirlos de cerca. Recientemente, un metanálisis de 12 estudios y 6.529 pacientes mostró que la CCC se asocia con una sustancial mejora de la supervivencia5. Este estudio demostró que los pacientes con alto grado de colateralización muestran una reducción significativa del riesgo de muerte en comparación con los pacientes con baja colateralización (riesgo relativo=0,64; IC95%, 0,45-0,91; p=0,012).
La urocinasa, que induce una cascada proteolítica local de gran importancia tanto en la remodelación vascular como en la angiogénesis, es uno de los participantes clave en el mecanismo de respuesta a la lesión cardiovascular y el crecimiento colateral, lo que la convierte en una interesante y prometedora diana terapéutica en las enfermedades vasculares16,18,31. En modelos de remodelación vascular inducidos por flujo, el contenido de uPA se correlacionó con el nuevo crecimiento de la íntima32 y los estudios en tejido arterial de ratones transgénicos17 y de primates33 van en el mismo sentido. En un modelo murino de isquemia del miembro posterior, se ha comprobado que los ratones deficientes en uPA muestran menos capacidad endógena para restaurar el flujo sanguíneo que los ratones control19. Por otro lado, Traktuev et al34 demostraron que la sobreexpresión de uPA estimula el crecimiento de los vasos y la perfusión de los tejidos, lo que limita el daño miocárdico y ayuda a la posterior remodelación tras el infarto.
En la superficie celular, uPA, que es una proteína multifuncional multidominio, se une al receptor de alta afinidad de la urocinasa (uPAR)35, que se localiza en el borde de ataque de la célula migratoria, lo que permite la proteolisis regulada de proteínas de la matriz extracelular en la dirección de movimiento de la célula. Por otro lado, el complejo uPA-uPAR también regula la remodelación vascular por vías independientes de la proteolisis y la generación de plasmina: interacciona, entre otros, con proteínas de matriz, integrinas y receptores de endocitosis, y activa la señalización intracelular y la expresión génica, con lo que regula la adhesión celular, la migración, la diferenciación y la proliferación16. Por otra parte, la arteriogénesis también se promueve por la infiltración de leucocitos mediada por uPA que no depende de uPAR19.
Respecto al SNP PLAU P141L, se ha demostrado que la sustitución de prolina por leucina en la posición del aminoácido 121 aumenta la hidrofugacidad de la región21. Esta sustitución puede afectar también a la estructura terciaria de la molécula, ya que la P121 es un componente de una de las tres estructuras de hoja beta antiparalelas en el dominio kringle de uPA21. Este cambio puede ser causa directa o indirecta de que disminuya la afinidad aparente por los coágulos de fibrina.
La capacidad de uPA para inducir quimiotaxis en las células musculares lisas se ha atribuido a la unión del dominio kringle de uPA a la superficie de las células y la asociación de uPA con uPAR36. El uPA puede unirse simultáneamente a dos receptores en la superficie celular: a uPAR a través del dominio semejante al factor de crecimiento y a la integrina Mac-1 a través de los dominios kringle y proteolítico37. Este complejo trimolecular multicontacto puede desempeñar un papel importante en el control de la migración de células inflamatorias y en la homeostasis vascular37. Por otra parte, el dominio kringle de uPA se une en la superficie celular a un receptor específico que es distinto de uPAR36 y contiene una secuencia que interactúa con el inhibidor del activador del plasminógeno tipo 138. El dominio kringle está implicado en la señalización intracelular y en la inducción de la migración y la adhesión celulares39.
Además de la asociación de la variante T de uPA con una reducción de la CCC presentada en este trabajo, distintos estudios demuestran que el SNP PLAU P141L está involucrado en otros estados patológicos, como los trastornos neurológicos y el cáncer. De particular interés es el papel de uPA en la enfermedad de Alzheimer20,23,25,26,40,41. Este es atribuible a su implicación funcional en la generación de plasmina, proteasa capaz de degradar las proteínas betaamiloides. Cabe destacar que Riemenschneider et al23 encontraron mayores recuentos de placas en las cortezas temporales de los pacientes portadores del alelo T de PLAU P141L en comparación con los pacientes sin este alelo, y que un reciente metanálisis ha confirmado que el alelo T incrementa el riesgo de tener esta enfermedad26. En otras enfermedades como el cáncer, se ha podido observar una asociación entre el genotipo CC de este polimorfismo y la invasión y metástasis del cáncer colorrectal22 o la relación del SNP con la susceptibilidad al carcinoma escamoso de lengua27. Además, Begin et al24 observaron una correlación de este SNP con la función de uPA a través de la matriz extracelular en la patogenia del asma. Muy recientemente, se ha publicado una asociación del SNP P141L con las concentraciones de lípidos en suero28 y, finalmente, con la enfermedad inflamatoria intestinal29.
Estas observaciones ponen de relieve la importancia biológica de este SNP funcional de uPA, que afecta a la estructura kringle de la proteína, y son coherentes con la asociación entre el alelo T en pacientes con EAC y la CCC reducida detectada en esta subpoblación.
Estas observaciones confirman la implicación del SNP PLAU P141L en el proceso arteriogénico de crecimiento de las arteriolas coronarias colaterales. Es posible que la variante L141, que se ha demostrado que afecta a la estructura del dominio kringle de la proteína y la posterior reducción de su afinidad por la fibrina, pueda afectar a la eficiencia de la remodelación de la matriz extracelular y la capacidad de migración de las células musculares lisas, con lo que disminuye la respuesta arteriogénica de los pacientes con EAC portadores de la variante alélica T.
CONCLUSIONESLos resultados presentados muestran, por primera vez, una fuerte asociación entre el SNP PLAU P141L y la CCC. Los pacientes con EAC con la variante funcional L141 muestran mayor probabilidad de tener una CCC deficiente. Aunque los resultados presentados en este documento deben replicarse en cohortes adicionales, el polimorfismo PLAU P141L podría ser una herramienta genética predictora de la respuesta arteriogénica en los pacientes con EAC. Estudios basados en el análisis genético de grandes muestras de casos y controles mediante genotipificación de miles de SNP distribuidos por todo el genoma (estudios de asociación de genoma completo) permitirán descubrir otros loci asociados con el desarrollo de CCC, como recientemente se ha demostrado para la enfermedad coronaria y el infarto de miocardio (revisado por Companioni et al42. en 2011). Además, dado el papel central de uPA en la formación de la neoíntima durante la remodelación arterial, se requieren ensayos de migración in vitro utilizando células musculares lisas de los pacientes que poseen el alelo T para comprobar la relevancia funcional de este polimorfismo. Por otra parte, y como defienden distintos autores16, estos resultados respaldan la idea de que la terapia génica de uPA o la mejora de la producción local de uPA podrían ser herramientas prometedoras para la estimulación local del crecimiento arterial colateral y el tratamiento del miocardio isquémico.
FINANCIACIÓNEste trabajo recibió el apoyo de la Universitat de Barcelona (proyecto ACESBELL 08) y la Fundació La Marató de TV3 07 (proyecto 080810).
CONFLICTO DE INTERESESNinguno.
Damos las gracias a Àlex Cordero, Montse Cairó, Eva Sánchez, Dolors Colell, Teresa Torrent, María José Fernández de Muniain y Miquel Rugat por su colaboración y asistencia técnica.