Fabry disease (FD) is an X-linked disorder caused by mutations in the GLA gene. This leads to deficiency in α-galactosidase A, the enzyme responsible for degrading certain glycosphingolipids. The enzyme deficiency leads to accumulation of these compounds, in turn resulting in dysfunction of vital organs (mainly the kidney and heart) and premature death. Both sexes are affected in similar proportions, but the disease is usually less serious and presents later in life in women. FD is classed as a rare disease, with a frequency of between 1:40 000-1:120 000.1
Diagnosis is challenging and cardiologists are often the first to suspect the disease after patients are referred for investigation of hypertrophic cardiomyopathy (HCM). The diagnosis is confirmed by genetic study, which can prompt a family study; however, there are genetic variants of doubtful pathogenicity.1 Pathogenicity is, however, a crucial consideration as enzyme replacement therapy (ERT) is available but is expensive and not free of complications.
We present the case of a family with FD in which the p.Arg118Cys variant was demonstrated not to cause the disease, thus highlighting the need to be alert to other similar variants.
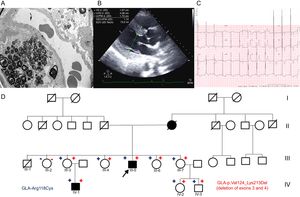
A 45-year-old man with proteinuria since he was 36 years old and whose renal function was deteriorating underwent a kidney biopsy. Electron microscopy showed zebra bodies indicative of FD (Figure A). In view of these findings, the patient was referred to the hereditary heart disease unit to complete the study. Echocardiography showed severe left ventricular hypertrophy (LVH) and electrocardiography revealed short PR interval and LVH (Figure B and C). A genetic study was conducted by next-generation sequencing of a panel of 16 genes associated with HCM (including GLA). The p.Arg118Cys variant was detected, but deletion of exons 3 and 4 (p.Val124_Lys213del) was also present.

A: Electron microscopy of kidney biopsy with zebra bodies. B: Echocardiogram of the index case with LVH. C: Electrocardiogram of index case with LVH and short PR interval. D: Family tree; square, male; circle, female; in boldface, phenotype involvement; arrow, proband; diagonal, dead;+red, carrier of exon 3 and 4 deletion; -red, noncarrier of exon 3 and 4 deletion;+blue, carrier of p.Arg118Cys variant; blue, noncarrier of the p.Arg118Cys variant. LVH, left ventricular hypertrophy.
The family study (Figure D) found that the patient's mother also had HCM and had died of kidney failure at age 64 years. Some family members were living in another region of Spain, and we therefore contacted another hereditary heart disease unit. Five female and 2 male carriers were detected, 1 of them with a 4-year-old boy (the clinical characteristics are summarized in the Table). All carriers of the GLA deletion are also carriers of the p.Arg118Cys variant, whereas noncarriers are not. The family study established that both the missense variant and the deletion were on the same allele (Figure 1A).
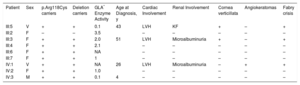
Clinical and Genetic Data of the Family Studied
| Patient | Sex | p.Arg118Cys carriers | Deletion carriers | GLA* Enzyme Activity | Age at Diagnosis, y | Cardiac Involvement | Renal Involvement | Cornea verticillata | Angiokeratomas | Fabry crisis |
|---|---|---|---|---|---|---|---|---|---|---|
| III:5 | V | + | + | 0.1 | 43 | LVH | KF | + | – | + |
| III:2 | F | – | – | 3.5 | – | – | – | – | – | |
| III:3 | F | + | + | 2.0 | 51 | LVH | Microalbuminuria | + | – | + |
| III:4 | F | + | + | 2.1 | – | – | – | – | – | |
| III:6 | F | + | + | NA | – | – | – | – | – | |
| III:7 | F | + | + | 1 | – | – | – | – | – | |
| IV:1 | V | + | + | NA | 26 | LVH | Microalbuminuria | – | + | + |
| IV:2 | F | + | + | 1.0 | – | – | – | – | – | |
| IV:3 | M | + | + | 0.1 | 4 | – | – | – | – | – |
–, no involvement; +: involvement; F, female; Fabry crisis, urent pain in hands and feet; KF, kidney failure; LVF, left ventricular hypertrophy; M, male; NA, not available;
Similar deletions that do not appear in the databases of normal populations had already been reported in association with FD. The deletion arises from reordering of repetitive sequences denominated Alu, which are distributed throughout the entire gene. If this variant were translated, part of the domain where the substrate binding site is located would be lost, and so the protein would lack any enzyme activity.
It was previously reported that p.Arg118Cys was responsible for FD,2 but studies do not confirm family cosegregation and provide information only on the index case. Furthermore, these studies are of the Sanger type, and therefore it is not possible to rule out that undetected deletions were causing the disease with this method. This limitation is overcome with next-generation sequencing with the platform that includes study of copy number variations.
The p.Arg118Cys variant also appears in the normal population: 8 hemizygous men were identified from a control population in the Exome Aggregation Consortium database.3 The presence of the variant in genotype databases of the general population with a high frequency (in excess of the prevalence of FD in the general population) supports the lack of pathogenicity of this mutation. Ferreira et al.4 showed that carriers of the p.Arg118Cys variant have similar survival to the control population and do not experience the complications of FD. It is important to highlight that enzyme activity could be reduced, but residual activity is probably sufficient for the enzyme to exercise its function, and therefore this measure should not be used to define pathogenicity and it certainly should not be used as a criterion for initiating ERT.
In the family studied, the index case was receiving ERT, and this treatment was offered to 1 nephew (IV:1) and 1 sister (III:2). The remaining female carriers were asymptomatic and showed no signs of the disease. The 4-year-old boy is asymptomatic but is being closely monitored, given the possibility of starting ERT early to avoid damage to target organs.
In summary, FD should include differential diagnosis with HCM and, if indicated, genetic study should be performed with next-generation sequencing to detect deletions. The pathogenicity of some variants previously considered pathogenic should be questioned in cases such as p.Arg118Cys, if deletions may have gone undetected and familial cosegregation was not confirmed. These issues can be overcome by study of the entire family and appropriate genetic study.5
CONFLICTS OF INTERESTL. Monserrat-Iglesias is a shareholder of Health in Code S.L.
.