Keywords
INTRODUCTION
The single most important complication in hypertrophic cardiomyopathy (HCM) is sudden death. Although in most patients sudden death is caused by ventricular arrhythmias (sustained ventricular tachycardia or ventricular fibrillation [VF]), it can also be caused by cardiac conduction disturbances (CCDs).1 Many studies report on the prevalence, determining factors and prognostic value of supraventricular or ventricular tachyarrhythmias, but practically no data exist on the prevalence and relevance of severe conduction disturbances in patients with HCM.2,3
The objectives of the present study are to analyze the prevalence of CCDs leading to permanent pacemaker (PM) implantation in a broad-ranging cohort of patients with HCM and to describe permanent PM recipient characteristics and outcomes.
METHODS
Retrospective study of permanent PM recipients with implantation indicated for bradycardia or symptomatic CCDs in a cohort of adult patients with HCM under observation since 1995 in a specialist clinic. Diagnosis of HCM was based on the presence of a left ventricle with ≥1.5 cm maximum parietal thickness in the absence of illness capable of causing the hypertrophy observed. In patients with a family history of HCM, we established ≥1.3 cm thickness and/or electrocardiogram (ECG) alterations compatible with HCM as diagnostic criteria.4,5
The protocol included ≥1 clinical examination per year, family tree, ECG, echocardiogram, Holter and ergometry. We took a sample of peripheral blood for genetic study (after informed consent). We used a diagnostic platform including the >600 HCM-related mutations described to date. We performed a discriminatory test for Fabry disease (FD) to determine alpha-galactosidase A (AGA) enzyme activity in blood. If storage disease was suspected, we performed clinical and analytical tests for lesions in other organs.
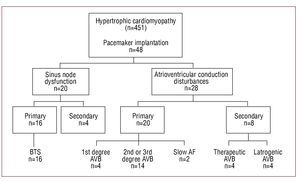
Indications for PM implantation were classified as:
1. Sinus node dysfunction (SND):
- Primary SND: bradycardia-tachycardia syndrome (BTS).
- Secondary SND: sinus bradycardia secondary to drugs (used in treating tachyarrhythmias and/or subvalvular aortic obstruction).
2. Atrioventricular (AV) conduction disturbances:
- Primary atrioventricular block (AVB): first degree; second and third degree; slow atrial fibrillation (AF).
- Secondary AVB: due to therapeutic ablation intended to treat supraventricular arrhythmias; iatrogenic AVB, linked with myectomy or septal ablation.
Causes of death were classified as cardiac or noncardiac according to clinical case history records.
RESULTS
Among 451 patients with HCM (64% men; mean age, 53 years; mean follow-up, 5.2 [1.3] years), 48 required permanent PM implantation (11%). Patient characteristics are in Table 1.

We found a family history of HCM in 23 patients (48%); of sudden death in 11 (23%) and of PM implantation in 9 (18%).
Method of Stimulation
The method of stimulation chosen was DDDR (30), VVIR (16), and AAI (1, changed to VVIR at 8 years). In one patient indicated for permanent PM we implanted a defibrillator (ICD) for primary prevention due to the risk factors presented. In 5 PM recipients, these were replaced by ICDs during follow-up for primary prevention.
Indications for Permanent Pacemaker Implantation (Figure)

Figure. Indications for permanent pacemaker implantation. AF indicates atrial fibrillation; AVB, first, second and third degree atrioventricular block; BTS, bradycardia-tachycardia syndrome.
Pacemaker implantation was indicated for primary bradyarrhythmia in 36 patients (75%) and for causes secondary to therapeutic regimens in 12 (25%).
In patients with PM we performed 8 interventions that entailed risk or potential risk of complete AVB. We performed 4 AV node ablations and subsequent PM implantation in patients with fast AF and poor response to medical treatment, and 4 PM implantations following myectomies.
Events During Follow-up
In a mean 5.9 years of follow-up, events occurred in 10 patients (2 transplantations and 8 deaths: 3 from heart failure, 1 kidney failure, 1 liver failure, 1 of neogastric origin and 1 unknown cause). Total mortality (all-cause death or transplantation) was 3.4% per year.
Genetic Study
We identified mutations in 15 patients (31%) (Table 2). In 1 patient with low AGA activity we found a mutation of the GLA gene (Galactosidase alpha) linked with FD. In another, with apical HCM, a mutation of the PKP2 (plakophilin-2) gene was linked with the development of arrhythmogenic right ventricular dysplasia (ARVD). In this case, we looked for mutations linked with the development of ARVD because the patient's father was the carrier of a cardiomyopathy compatible with this phenotype.

DISCUSSION
In HCM, slow rhythm has been considered an uncommon and AVB an infrequent complication. However, isolated cases have been reported in which HCM presentation was due to CCDs,6-8 and some series describe PRKAG2 gene mutations associated with HCM, Wolf-Parkinson-White syndrome and CCDs.9 In some of these cases, authors suggest a possible family history of CCDs.10-14
However, in HCM, despite being the cardiomyopathy most frequently found in the general population, no systematic studies of CCDs and permanent PM implantation have been published. Most published reviews of HCM and PM refer to therapeutic PM use (to treat subaortic obstruction and associated symptoms) or to PMs in the treatment of complications arising from other therapeutic procedures (such as myectomy or septal ablation).1,2 In our series, we found a high prevalence of CCDs leading to PM implantation (11%). If we discount patients who required PM implantation for SND secondary to drugs, iatrogenic AVB, or therapeutic AV node ablation, 36 (8%) patients experienced CCDs that required permanent PM implantation. The population in question is relatively young (mean, 58 years), symptomatic (syncope, 44%) and most had conserved ventricular function (89%).
Both functional level and cardiovascular mortality were greater than in the series reported by Elliott et al14 (0.5%) in a cohort of 956 patients with HCM. Our series included a higher percentage of patients in New York Heart Association (NYHA) functional classes III and IV (42% vs 3% reported by Elliott et al) and cardiovascular mortality was 2.1% per year, versus 0.5%. These differences are probably explained by the fact that our patients with HCM and PM were older (mean, 58 [13] years) than those reported by Elliott et al (42 [15] years) and also because CCDs can be a marker of seriousness in HCM.
In our series, 18% of patients had at least one (first or second degree) family member who was a PM recipient. We have identified one single mutation, E101K in the cardiac actin gene, in 3 index cases (PM recipients) in families that initially were not considered related and that may have a common origin. Our study shows the existence of a family history of CCDs in HCM patients. In turn, this shows these disturbances can form part of the phenotype expression of HCM. The clinical implications of this are obvious: in the diagnosis and follow-up of patients with HCM, we should be alert to the presence of (supraventricular or ventricular) tachyarrhythmias, and look for and expect CCDs to have occurred or to occur in the future.
Limitations
This is a retrospective study in which indications for permanent PM implantation have not been established a priori and some implantations have been conducted in other centers; both are factors that may bias indications for implantation. However, we are dealing with a symptomatic population largely needing treatment with non-chronotropic drugs and that, due to the presence of prior bradyarrhythmias/ tachyarrhythmias, underwent permanent PM implantation.
The genetic study was incomplete because we only studied previously described mutations in the principle genes involved, making it impossible to establish potential genotype-phenotype correlations: we have not studied the LAMP2 gene and have only studied some mutations of the PRKAG2 gene, both of which are related with cardiac hypertrophy and CCDs.
CONCLUSIONS
In our series of patients with HCM we found a high incidence of CCDs leading to permanent PM implantation. In this group, family history is evident (18%). Cardiac conduct disturbances seem to form part of the phenotype expression of HCM. We need new studies to confirm this association and genotypephenotype studies to clarify the link between some mutations and CCDs.
Dr Xusto Fernández-Fernández receives financial support from the RECAVA Cardiovascular Research Network of the Instituto Carlos III, Madrid. Dr Martin F. Ortiz receives financial support from the BBVA-Carolina Foundation. This study received financial support from project FIS 2005: PI050377, of the Instituto Carlos III, Madrid.
Correspondence: Dr. R. Barriales Villa.
Instituto de Investigación Biomédica A Coruña (INIBIC). Hospital Marítimo de Oza.
As Xuvias, s/n. 15006 A Coruña. España.
E-mail: rbarrialesv@gmail.com
Received March 17, 2009.
Accepted for publication August 6, 2009.