Palabras clave
INTRODUCCIÓN
La complicación más importante en la miocardiopatía hipertrófica (MCH) es la muerte súbita. A pesar de que en la mayoría de los pacientes la causan las arritmias ventriculares (taquicardia ventricular sostenida o fibrilación ventricular [FA]), también puede producirse por trastornos de la conducción cardiaca (TSC)1. Mientras que existen múltiples estudios sobre la prevalencia, los determinantes y el valor pronóstico de las taquiarritmias (supraventriculares o ventriculares), prácticamente no hay datos sobre la prevalencia y la relevancia de los trastornos graves de la conducción en pacientes con MCH2,3.
Los objetivos de este trabajo son el análisis de la prevalencia de TSC que determinaron el implante de marcapasos (MP) en una amplia cohorte consecutiva de pacientes con MCH y la descripción de las características y la evolución de los pacientes a los que se implantó un MP.
MÉTODOS
Estudio retrospectivo de pacientes portadores de MP implantados por bradicardia o TSC sintomáticos en una cohorte de pacientes adultos con MCH controlados desde el año 1995 en una consulta especializada. El diagnóstico de MCH se realizó por la presencia de un ventrículo izquierdo con espesor parietal máximo 3 1,5 cm en ausencia de enfermedad capaz de producir la hipertrofia observada. En pacientes con antecedentes familiares (AF) de MCH, se consideró diagnóstico un grosor 3 1,3 cm y/o las alteraciones electrocardiográficas compatibles con MCH4,5.
El protocolo incluye evaluación clínica al menos anual, árbol familiar, ECG, ecocardiograma, Holter y ergometría. Se extrajo sangre periférica para estudio genético (tras consentimiento informado). Se empleó una plataforma diagnóstica que incluía más de 600 mutaciones relacionadas con MCH descritas hasta el momento. Se realizó criba de enfermedad de Fabry (EF) evaluando en sangre la actividad de la enzima alfagalactosidasa A (EAGA). Ante la sospecha de enfermedad de depósito, se realizaron valoraciones clínicas y analíticas de afección de otros órganos.
Las indicaciones de implante de MP se clasificaron en:
1. Disfunción del nodo sinusal (DNS):
- DNS primario: síndrome bradicardia-taquicardia (SBT).
- DNS secundario: bradicardia sinusal secundaria a fármacos (usados para el tratamiento de las taquiarritmias y/o la obstrucción subvalvular aórtica).
2. Trastornos de la conducción auriculoventricular (AV):
- Bloqueo auriculoventricular (BAV) primario: de primer grado; de segundo y tercer grado; FA lenta.
- BAV secundario: por ablación terapéutica con intención de tratar las arritmias supraventriculares; BAV iatrogénico, vinculado a miectomía o ablación septal.
En cuanto a las causas de muerte, fueron catalogadas como cardiacas o no cardiacas según la información documentada en la historia clínica.
RESULTADOS
De un total de 451 pacientes con MCH (el 64% varones; media de edad, 53 años; seguimiento medio, 5,2 ± 1,3 años), requirieron implante de MP 48 (11%). Las características se resumen en la tabla 1. Se describieron AF de MCH en 23 pacientes (48%); en 11 (23%) había AF de muerte súbita y en 9 (18%), AF de implante de MP.

Modo de estimulación
El modo de estimulación seleccionado fue DDDR (30), VVIR (16) y AAI (1, cambiado por VVIR a los 8 años). En un paciente con indicación de MP se implantó un desfibrilador (DAI) por prevención primaria debido a los factores de riesgo que presentaba, y en 5 casos en los que se había implantado un MP, éste fue sustituido por un DAI durante el seguimiento por prevención primaria.
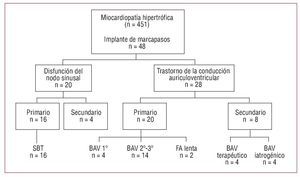
Indicaciones de implante de marcapasos (fig. 1)

Fig. 1. Indicaciones de implante de marcapasos. BAV: bloqueo auriculoventricular de primero, segundo y tercer grado; FA: fibrilación auricular; SBT: síndrome bradicardia-taquicardia.
Las bradiarritmias primarias fueron causa de implante de MP en 36 pacientes (75%), mientras que las causas secundarias a alguna de las terapias aplicadas a estos pacientes se presentaron en 12 casos (25%).
En los pacientes con MP se realizaron 8 procedimientos intervencionistas que conllevan riesgo de BAV completo o lo buscan. Así, se realizaron 4 ablaciones del nodo AV y posterior implantación de MP, por FA rápidas con mala repuesta a tratamiento médico, y 4 implantaciones de MP tras miectomías.
Eventos durante el seguimiento
En un seguimiento medio de 5,9 años se produjeron eventos en 10 pacientes (2 trasplantes y 8 muertes: 3 por insuficiencia cardiaca, 1 por insuficiencia renal, 1 por insuficiencia hepática, 1 por neogástrica y 1 de causa desconocida). La mortalidad total (muerte por todas las causas o trasplante) fue del 3,4% por año.
Estudio genético
Se identificaron mutaciones en 15 pacientes (31%) (tabla 2). En un paciente con actividad baja de la EAGA se encontró una mutación en el gen GLA (alfagalactosidasa A) vinculada con EF. En otro, con MCH apical, una mutación en el gen PKP2 (plakofilina-2) vinculada con el desarrollo de displasia arritmogénica de ventrículo derecho (DAVD). En este caso se buscaron mutaciones vinculadas con el desarrollo de DAVD porque el padre del paciente era portador de una miocardiopatía compatible con ese fenotipo.

DISCUSIÓN
Se ha considerado que los ritmos lentos son complicaciones poco comunes en la MCH y que el BAV es una complicación poco frecuente. No obstante, se han publicado casos aislados en los que la forma de presentación de la MCH fue por TSC6-8, así como alguna serie que describe mutaciones en el gen PRKAG2 relacionadas con MCH, síndrome de Wolf-Parkinson-White y TSC9. En alguno de esos casos se señala la posible agregación familiar de los TSC10-14.
Sin embargo, en la MCH, pese a ser la miocardiopatía descrita en población general más frecuentemente, no hay estudios sistemáticos publicados sobre TSC e implante de MP. La mayoría de las revisiones publicadas sobre MCH y MP hacen referencia al uso de éstos desde el punto de vista terapéutico (para el tratamiento de la obstrucción subaórtica y de los síntomas asociados con ella) o como tratamiento de las complicaciones producidas por otros procedimientos terapéuticos (como las miectomías o las ablaciones septales)1,2. En nuestra serie hemos encontrado una alta prevalencia de TSC que llevaron a la implantación de MP (11%). Si no consideramos a los pacientes que requirieron MP por DNS secundaria a fármacos o por BAV iatrogénico o por ablación terapéutica del nodo AV, fueron 36 (8%) los pacientes que sufrieron algún TSC que requirió el implante de MP. Se trata de una población relativamente joven (media, 58 años), sintomática (síncope, 44%) y en su mayor parte con función ventricular conservada (89%).
Si consideramos el grado funcional y la mortalidad cardiovascular, ambos fueron superiores a los declarados por Elliott et al14 (0,5%) en su cohorte de 956 pacientes con MCH; así, en nuestra serie hay mayor porcentaje en grado funcional III y IV de la New York Heart Association (NYHA) (el 42 frente al 3% de Elliott) y la mortalidad cardiovascular fue del 2,1% al año, frente al 0,5%. Estas diferencias probablemente se deban a que nuestros pacientes con MCH y MP tienen una media de edad mayor (58 ± 13 años) que la serie de Elliott (42 ± 15 años), pero también a que los TSC puedan ser un marcador de severidad en la MCH.
El 18% de los pacientes de nuestra serie tenían al menos un familiar (de primero o segundo grado) portador de MP. Así, hemos identificado una misma mutación, la E101K en el gen de la actina cardiaca, en 3 casos índice (portadores de MP) de familias que inicialmente se consideró no relacionadas y que posiblemente tengan un origen común. Nuestro estudio muestra la existencia de una agregación familiar de los TSC en la MCH. Nos indica a su vez que estos trastornos pueden formar parte de la expresión fenotípica de la MCH. Las implicaciones clínicas de este hecho son evidentes: en el diagnóstico y el seguimiento de los pacientes con MCH, no sólo debemos estar alerta ante la presencia de taquiarritmias (supraventriculares o ventriculares), sino que debemos buscar y esperar que haya TSC o vayan a producirse.
Limitaciones
Se trata de un estudio retrospectivo en el que las indicaciones de implantación de MP no han sido establecidas a priori, e incluso alguna se ha llevado a cabo en otros centros hospitalarios, con lo que pueden introducirse sesgos en las indicaciones de implantación. De todos modos, se trata de una población sintomática que en gran parte precisó tratamiento con fármacos no cronotrópicos y a la que, debido a la presencia de bradiarritmias/taquiarritmias previas, se implantó MP.
En cuanto al estudio genético, éste no ha sido completo, ya que sólo se han estudiado algunas mutaciones (ya descritas) en los principales genes implicados, por lo que no se ha podido establecer posibles correlaciones genotipo-fenotipo. De esta manera no se ha estudiado el gen LAMP2 y sólo se han estudiado algunas mutaciones en el gen PRKAG2, ambos relacionados con la hipertrofia cardiaca y los TSC.
CONCLUSIONES
En nuestra serie de pacientes con MCH encontramos una elevada incidencia de TSC que determina el implante de MP. En este grupo hay una agregación familiar evidente (18%). Los TSC parecen formar parte de la expresión fenotípica de la MCH. Se necesitan nuevos estudios que confirmen esta asociación y estudios genotipo-fenotipo que aclaren la vinculación de algunas mutaciones con los TSC.
Full English text available from: www.revespcardiol.org
Financiación: el Dr. Xusto Fernández-Fernández recibe financiación de la Red de Investigaciones Cardiovasculares (RECAVA) del Instituto Carlos III de Madrid; el Dr. Martín F. Ortiz recibe financiación de la Fundación Carolina-BBVA. Este estudio ha recibido financiación del proyecto FIS 2005: PI050377, Instituto Carlos III de Madrid.
Correspondencia: Dr. R. Barriales Villa.
Instituto de Investigación Biomédica A Coruña (INIBIC). Hospital Marítimo de Oza.
As Xuvias, s/n. 15006 A Coruña. España.
Correo electrónico: rbarrialesv@gmail.com
Recibido el 17 de marzo de 2009.
Aceptado para su publicación el 6 de agosto de 2009.