Keywords
INTRODUCTION
Pulmonary atresia with intact ventricular septum (PAIVS) is the complete obstruction of the right ventricular outflow tract due to pulmonary valve atresia in the absence of any ventricular septal defect. Pulmonary blood flow is therefore dependent on a patent ductus arteriosus.1 The association of PAIVS with aortic stenosis is highly unusual and only a very few cases have been reported.
We present the case of a 19-year-old primiparous woman whose fetal echocardiogram at 24 weeks of gestation showed PAIVS. The neonate had desaturation and an aortic systolic ejection murmur. Echocardiogram showed PAIVS and a thickenedand stenotic bicuspid aortic valve. The neonate suffered low cardiac output which could not be controlled, and died.
CLINICAL CASE
A 19-year-old primiparous woman with no medical history of note underwent fetal echocardiogram which showed PAIVS, a hypoplastic right ventricle and color Doppler images suggesting coronary sinusoids. Delivery was at term with prostaglandin-E1 infusion at 0.03 µg/kg/min. At birth there was mild desaturation, a hyperdynamic heart beat, thrill and a III/VI aortic systolic ejection murmur. Blood pressure was 50/32 mm Hg and heart rate was 148 bpm. A chest x-ray revealed moderate cardiomegaly, and an electrocardiogram (ECG) showed sinus tachycardia, axis +60º, P pulmonale, decreased right ventricular voltage and signs of left ventricular systolic overload (Figure 1). Echocardiography at 2 hours of life (Figure 2) showed PAIVS, hypoplasia of the tricuspid valve and the body and infundibulum of the right ventricle, sinusoids, a patent ductus arteriosus, a thickened stenotic bicuspid aortic valve (with a maximum gradient of 45-50 mm Hg) and a dilated and hypertrophied left ventricle. Pulmonary valvulotomy with a radiofrequency catheter, systemic-to-pulmonary shunt, and aortic valvulotomy were considered. However, the patient's condition worsened steadily, with low cardiac output which could not be controlled, and she died at four days of age. Autopsy study (Figure 3) confirmed the echocardiographic findings.

Fig. 1. Initial complementary studies. A: electrocardiogram of the patient at 2 hours of life showing sinus tachycardia, electrical axis 60º, P pulmonale, reduced right ventricular voltage and signs of left ventricular systolic overload. B: chest x-ray at 2.5 hours of life showing moderate cardiomegaly.

Fig. 2. Echocardiogram at 2 hours of life. A: subcostal plane showing a thickened aortic valve (arrow). B: parasternal short axis showing pulmonary valve atresia (arrow) with dilation of the trunk of the pulmonary artery (TP). LA indicates left atrium; Ao, aortic valve; RV, right ventricle; LV, left ventricle.

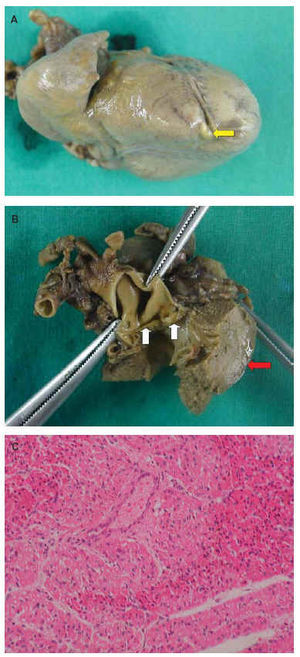
Fig. 3. Pathological study. A: gross image of the patient's heart with a sinusoid (arrow) at the apex of the right ventricle beside the interventricular septum. B: gross image showing thickening of the 2 leaflets of the aortic valve (yellow arrows) and marked left ventricular hypertrophy (red arrow). C: microscopic image of the papillary muscle of the left ventricle. Hematoxylin-eosin. There are signs of patchy ischemia (left and right top edge) with more eosinophilic cytoplasm, and hyperchromatic and pyknotic nuclei.
DISCUSSION
Pulmonary atresia with intact ventricular septum is uncommon, occurring in only 0.7% of the patients with congenital heart disease. It involves complete obstruction of the right ventricular outflow tract as a result of pulmonary valve atresia, typically with fusion of the three commissures. There is no association with any interventricular septal defect. A patent ductus arteriosus is required for pulmonary blood flow.1 The disease is accompanied by various other morphological anomalies. The right ventricle may be small, normal sized or dilated (with thin walls and associated tricuspid valve incompetence). Patients in whom the right ventricle is small have hypoplastic right heart syndrome (HRHS), characterized by PAIVS and a hypoplastic tricuspid valve, with a poorly developed ring and papillary muscles. When the right ventricle is normal sized or dilated, the tricuspid valve is almost normal sized but can be severely incompetent.2,3 The right atrium is dilated and may appear aneurysmal on echocardiography if tricuspid valve incompetence is present. Blood from the cardiac circulation leaves the right heart via a patent foramen ovale or another interatrial septal communication. The trunk and pulmonary branches are well formed. The left atrium is slightly or moderately dilated because it receives both the systemic and the pulmonary venous return. The left ventricle is enlarged and its distensibility and contractility are altered. As a result of the high pressure in the right ventricle, the embryonic connections of the ventricular chamber with the coronary circulation remain, forming the sinusoids, which are connected with the myocardial capillary bed and, via the latter, with the epicardial coronary arteries.4 The sinusoids carry poorly oxygenated blood to the myocardium and can end blindly.5 They are subjected to high systolic pressures which lead to hypertrophy of the middle layer of the intramural arteries6 and progressive enlargement of the sinusoids, which may result in myocardial rupture.2 A further consequence of sinusoids communicating with one or both coronary arteries is a phenomenon of ischemic steal from the coronary capillary bed during diastolic flow. Blood passes from the coronary arteries to the sinusoids, and finally to the right ventricle, and infarction can result.7 There may occasionally be stenosis in the sinusoids and an increased risk of ischemia and death because coronary artery circulation depends on the right ventricle.
Other abnormalities have been reported in the coronary arteries, such as fistulas1 and stenosis or atrophy.8 In this latter case, typical of hearts with right ventricular hypoplasia, the distal coronary vessels are replaced by communications with the right ventricle, upon which coronary vessel circulation therefore depends. In the absence of fetal development of severe tricuspid valve incompetence, there is no resulting congestive heart failure (with pleural and pericardial effusion and ascites) and the incidence of fetal death is low.9,10 The clinical manifestations in the newborn are cyanosis, hypoxemia, metabolic acidosis, and right and left heart failure. Those exceptional patients who survive beyond the neonatal period without surgery are physically underdeveloped.2 Electrocardiogram shows normal or increased right ventricular voltages (in some patients with HRHS) and an enlarged right atrium. Chest x-ray shows a normal heart outline in patients with HRHS, whereas if the right ventricle is of normal size or dilated, dilatation of the right chambers can be seen.3 The prognosis is usually poor in the absence of treatment,11 with over 50% mortality at one month.2 Postnatal survival depends on the patency of the ductus arteriosus, which explains the requirement for continuous infusion of prostaglandin-E1; closure of the ductus would lead to death.5 Some patients have marked infundibular atresia which limits the success of pulmonary valvuloplasty. A high rate of survival can be achieved with surgery.12
The association of PAIVS with aortic stenosis is rare, with only a few cases reported, either in isolation13 or as part of HRHS.14 Survival, as in our patient, is unusual, possibly because of left ventricular failure from multifactorial causes. The alterations which can lead to this situation are myocardial ischemia and left ventricular dilation. The former may arise due to steal of blood from the coronary bed and the supply of poorly oxygenated blood from the right ventricle via the sinusoids, coronary hypoperfusion resulting from systemic hypoperfusion due to aortic stenosis, and reduced aortic diastolic blood pressure because of prostaglandin-E1 infusion. Left ventricular dilation may be due to the pressure overload caused by aortic stenosis.
Correspondence: Dr. J.R. Peraira.
San Marcial, 28, 1.o C. 28931 Móstoles. Madrid. España.
E-mail: robertoperaira@hotmail.com