We report 2 patients diagnosed with cardiomyopathy caused by a single genetic defect in the mitochondrial DNA. Both patients illustrate the importance of integrating clinical information when establishing a diagnosis, and allow us to discuss the unique characteristics of reproductive counselling for this type of genetic disease.
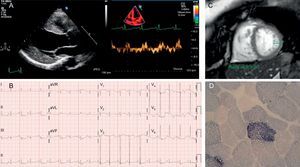
The first patient was a black Angolan man aged 47 years, who had moved to Spain to study ophthalmology. His clinical history included long-standing diabetes mellitus, recurrent tuberculosis, and bilateral sensorineural hearing loss. He was admitted for fever and acute respiratory failure. The echocardiogram (Figure) showed concentric left ventricular hypertrophy and severe systolic dysfunction. The cardiologic study was complemented with magnetic resonance imaging (absence of late-enhancement) and coronary angiography (normal). Other findings included marked cachexia, with steppage gait and renal failure with microalbuminuria. His mother had died of heart problems, his children were healthy, and a sister had an unspecified heart disease. Mitochondrial disease was suspected, specifically MIDD syndrome (maternally inherited diabetes and deafness) (Table). Sequencing of mitochondrial DNA from a blood sample revealed an m.3243A>G mutation in the MT-TL1 gene encoding mitochondrial tRNALeu, with 50% heteroplasmy. The patient was discharged with treatment for heart failure and returned to his home country.

A: Case 1 echocardiogram showing a dilated and hypertrophied left ventricle, with decreased tissue Doppler velocities. B: Case 1 electrocardiogram in sinus rhythm, with evidence of left ventricular enlargement. C: Cardiac magnetic resonance imaging of case 2, with increased trabeculation and noncompacted/compacted ratio >2.3. D: ragged red fibers with excess succinate dehydrogenase staining in case 2.
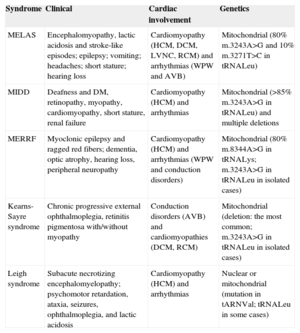
Principal Mitochondrial Syndromes Associated With the m.3243A>G Mutation in MT-TL1 that Present With Cardiac Involvement
| Syndrome | Clinical | Cardiac involvement | Genetics |
|---|---|---|---|
| MELAS | Encephalomyopathy, lactic acidosis and stroke-like episodes; epilepsy; vomiting; headaches; short stature; hearing loss | Cardiomyopathy (HCM, DCM, LVNC, RCM) and arrhythmias (WPW and AVB) | Mitochondrial (80% m.3243A>G and 10% m.3271T>C in tRNALeu) |
| MIDD | Deafness and DM, retinopathy, myopathy, cardiomyopathy, short stature, renal failure | Cardiomyopathy (HCM) and arrhythmias | Mitochondrial (>85% m.3243A>G in tRNALeu) and multiple deletions |
| MERRF | Myoclonic epilepsy and ragged red fibers; dementia, optic atrophy, hearing loss, peripheral neuropathy | Cardiomyopathy (HCM) and arrhythmias (WPW and conduction disorders) | Mitochondrial (80% m.8344A>G in tRNALys; m.3243A>G in tRNALeu in isolated cases) |
| Kearns-Sayre syndrome | Chronic progressive external ophthalmoplegia, retinitis pigmentosa with/without myopathy | Conduction disorders (AVB) and cardiomyopathies (DCM, RCM) | Mitochondrial (deletion: the most common; m.3243A>G in tRNALeu in isolated cases) |
| Leigh syndrome | Subacute necrotizing encephalomyelopathy; psychomotor retardation, ataxia, seizures, ophthalmoplegia, and lactic acidosis | Cardiomyopathy (HCM) and arrhythmias | Nuclear or mitochondrial (mutation in tARNVal; tRNALeu in some cases) |
AVB, atrioventricular block; CPEO, chronic progressive external ophthalmoplegia; DM, diabetes mellitus; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; LVNC, left-ventricular noncompaction; RCM, restrictive cardiomyopathy; MELAS, mitochondrial encephalopathy, lactic acidosis and stroke-like episodes; MERRF, myoclonic epilepsy with ragged red fibers; MIDD, maternally inherited diabetes and deafness; WPW, Wolff-Parkinson-White syndrome.
The second patient was a 36-year-old woman with multiple brain microbleeds identified during a study for hearing loss, and in whom cardiac evaluation revealed a possible noncompaction cardiomyopathy (Figure). The patient had had type 1 diabetes since the age of 24 years, and had short stature and low body mass index. There was no family history of heart disease, myopathy, or sensorineural problems. The patient reported frequent migraines. She was in New York Heart Association functional class II-III for exercise intolerance, had normal levels of creatine kinase and the amino terminal fraction of brain natriuretic peptide, and oscillating lactate levels (>2.5 mmol/L in several readings).
No further treatment was indicated, as ventricular function was normal. In this patient, the suspected clinical phenotype was MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) (Table). Muscle biopsy showed 8% ragged red fibers, mostly cyclooxygenase-positive (Figure), and the genetic study again revealed the m.3243A>G mutation in MT-TL1, with 87% to 91% heteroplasmy in the muscle biopsy.
Mitochondrial diseases are characterized by dysfunction of the respiratory chain, leading to cellular energy deficit. The most commonly affected organs are those with the highest metabolic demand, such as the nervous system and muscle. Common manifestations include encephalopathy, myopathy, diabetes, and hearing loss. The simultaneous involvement of several of these organs without an apparent common cause or manifestations early in life, such as diabetes or stroke before age 40 years, should serve as a guide to this diagnosis. These syndromes may present with widely different percentages of cardiac abnormalities (from 3%-81%, depending on the series) and in the form of both cardiomyopathies (usually hypertrophic or dilated) and conduction disorders (Table). The m.3243A>G mutation in mitochondrial tRNALeu is one of the most common, and can result in different syndromes such as MIDD (case 1) and MELAS (case 2), with considerable variation in cardiac manifestations.1 It is common to observe onset or worsening of symptoms after stressful situations (in the first patient, the hearing loss attributed to tuberculosis drugs had actually started years earlier, following an episode of malaria). In the presence of a classical clinical syndrome, the diagnosis can be confirmed by genetic analysis. Treatment is symptomatic, and should avoid drugs such as metformin (risk of lactic acidosis) or statins (worsening myopathy). Antioxidants or alternative therapies with coenzyme Q10 and L-carnitine are used, although there is controversy about their beneficial effects. Anesthesia should be used with special caution due to the risk of respiratory failure, with avoidance of nondepolarizing muscle relaxants and barbiturates.
Following diagnosis, the patient in case 2 expressed a desire to have children without this disease. Providing genetic counseling is one of the tasks facing cardiologists managing patients with familial heart disease.2
Mitochondrial genetics has a number of peculiarities to be taken into account when giving reproductive advice. Since zygote mitochondria come from the oocyte, inheritance is matrilineal, with women transmitting to all their descendants. The coexistence of more than one type of mitochondrial DNA molecule is called heteroplasmy. A minimum percentage of mutated DNA is necessary for symptoms to become evident (threshold effect). In cell division, the distribution of mitochondria is random, and daughter cells do not necessarily receive the same amount of mutated DNA. Mitochondrial DNA continues to replicate independently of cell division, such that initially healthy tissues may develop signs of disease over time. These features explain the phenotypic variation and clinical expression of these disorders, as well as the difficulty in preventing them by using assisted reproduction techniques. The reproductive options that guarantee the absence of disease transmission to offspring are adoption, gestation of an embryo from another couple, or fertilization of a donated egg. All have the disadvantage that the genetic link to the mother is lost.3
Preimplantation diagnosis for mitochondrial genetic disorders can only reduce the chances of disease by transferring embryos with low levels of heteroplasmy. Under current Spanish law, it is unlikely that this treatment would be authorized, since it does not ensure the use of embryos without genetic defects.4
Although still in the experimental stage, the future of prevention for these diseases lies in mitochondrial replacement techniques. These techniques consist of creating embryos with nuclear DNA from the parents, and mitochondrial DNA from a donor (“3-parent babies”). The efficacy and safety of these techniques is supported by a recent British scientific document supports, which calls for their translation to humans, in the current context in which it is not possible to use genetically modified embryos. Following publication of this document, and following popular consultation showing that public opinion was in favor of these techniques, the UK government has proposed to amend its legislation.5
The patient in case 2 finally decided to undergo in vitro fertilization with donated oocytes.
FUNDINGThis work was partially funded by ISCIII (PI11/0699 and RD12/0042/0066 to PGP and LAP; PI12/01795 to BB).