Left dominant arrhythmogenic cardiomyopathy (LDAC) exhibits characteristic phenotypic and genetic features which were found in the five Spanish family members described in this study. Triggered by a cold, a young man presented with a ventricular tachycardia of left ventricular origin and left ventricular late gadolinium enhancement. His resting ECG showed low potentials, delayed ventricular depolarization (inferior and V4-V6 leads) and atrioventricular conduction disturbances. His endomyocardial biopsy revealed myocyte loss with interstitial fibrosis. Despite the initial diagnosis of myocarditis, familial screening was pivotal in confirming the diagnosis of LDAC. A novel nonsense mutation in the desmoplakin gene (Q1866X) and the truncated protein which it produces were observed in skin samples.
Keywords
Arrhythmogenic cardiomyopathy (ACM) shows a prevalence of 1:5,000.1 Forty percent of cases are associated with mutations, the majority being found in genes encoding desmosomal proteins, which normally exhibit autosomal dominant inheritance1, 2 and variable penetrance. The remaining 60% are estimated to be linked to genes which have not yet been identified or to acquired causes.3
Desmosomes are responsible for ensuring cellular adhesion and are found in great numbers in tissues subjected to constant mechanical strain, such as the skin and the myocardium. The inclusion of defective proteins in the desmosomes of patients with ACM reduces their adhesive properties and encourages myocyte loss, replacement with fibro-fatty tissue and inflammation. These factors provide the anatomical substrate to generate ventricular tachyarrhythmias and sudden death.3
Originally described in the right ventricle (RV), a broader definition of ACM now also includes forms which primarily affect the left ventricle (LV) and are known as arrhythmogenic left ventricular cardiomyopathies (ALVC).4, 5, 6
We present the case of a Spanish family that was studied following an episode of sustained monomorphic ventricular tachycardia (SMVT) in the proband. The cardiological and genetic protocol diagnosed ALVC in the proband and a family member, and identified 2 other family members as genetic carriers and another subject as healthy. We will emphasize the characteristic features of this cardiomyopathy to make it easier to recognize and to add to databases with genotype-phenotype correlations.
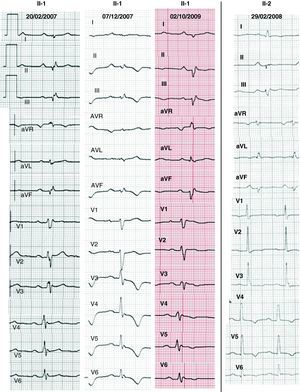
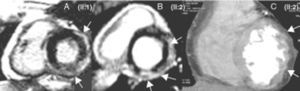
Methods PatientsThe proband, who was 36 years of age (Figure 1, II:1) and was seen as an outpatient, complained of atypical chest pain; an effort test, performed in accordance with the Bruce protocol, proved negative. His cardiovascular magnetic resonance (CMR) results suggested myocarditis, owing to late gadolinium enhancement (LGE) in the LV. He was admitted 1 year later with a cold and symptoms of dyspnea, syncope, and poorly tolerated SMVT, with morphology findings indicating right bundle-branch and superior axis block, which was cardioverted. The ECG showed low potentials, delayed ventricular depolarization (inferior and V4-V6 leads, and also occasionally V1-V3 leads) and atrioventricular conduction disturbances (Figure 2). The CMR coincided with the former findings, with evidence of slight LV systolic dysfunction. Wavy contours in the 64-slice multi-detector computed tomography (MDCT) images indicated myocardial infiltration by fatty deposits/epicardial fibrosis (Figure 3). An endomyocardial biopsy (EMB) revealed interstitial fibrosis and myocyte loss <30% with no fatty infiltration. The patient was fitted with a defibrillator and discharged.

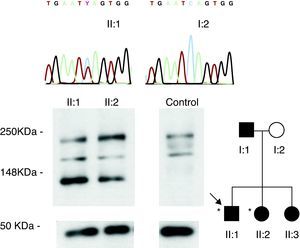
Figure 1. Family members included in the study (circles: females; squares: males; *left dominant arrhythmogenic cardiomyopathy phenotype). The chromatograms of the Q1866X mutation carriers (black symbols) and healthy subjects (white symbols) and their skin desmoplakin expression pattern are shown.

Figure 2. Electrocardiogram of patients with ACM showing low potentials, atrioventricular conduction disturbances, and delayed ventricular depolarization (inferior and V4-V6 leads, and also occasionally V1-V3 leads). The changes in repolarization (especially V4-V6) were intermittent in the proband.

Figure 3. A and B: cardiovascular magnetic resonance. C: multi-detector computed tomography. The arrows indicate late gadolinium enhancement (A and B) and myocardial infiltration as a result of fibrosis/epicardial fat deposition (C).
Although in this clinical study the proband did not meet ACM criteria,7 the fact that ALVC was suspected led us to evaluate the patient's family (his parents and two sisters) using standard ECG, echocardiography, effort test, 24-h Holter ECG, CMR, and general biochemical tests.
Having completed the family assessment, we screened the proband for mutations, conducting a family-based genetic cascade screening. A skin biopsy of the ALVC patients (II:1 and II:2) was then taken to analyze the pattern of desmoplakin expression in comparison with that of a control subject.
Genetic StudyDNA was extracted from peripheral blood leukocytes. The five main desmosomic genes (plakoglobin, plakophylin-2, desmoglein-2, desmocollin-2 and desmoplakin) were sequenced in both directions (ABI Prism 3100 sequencer, Applied Biosystems) in the proband. Once the mutation was identified, the affected exon was selectively sequenced in the family members and 200 control chromosomes.
Tissue AnalysisDesmoplakin expression in the affected subjects and a healthy control was analyzed by immunotransference in processed skin biopsies, as has been previously described,6 using Novex 4%-12% gel, Tris-Glycine 1 mm gel (Invitrogen) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the loading control. The primary antibodies were NW161 (desmoplakin N-terminal specific antibody, donated by Dr. Kathleen Green, Northwestern University, Evanston, Illinois, United States) and GAPDH rabbit mAb (14C10) (IZASA), and the secondary antibodies were sheep anti-mouse IgG-HRP (Amersham Bioscience) and rabbit anti-donkey IgG-HRP (Amersham Bioscience).
The protocol had previously been authorized by the ethics committee of our hospital and each subject signed an informed consent form.
Results ProbandThe genetic study identified a new variant of the desmoplakin gene (c. 5596C>T, Q1866X) (Figure 1), which was absent in controls and led to a stop codon in the translation in the central rod domain of the protein. Immunotransference detected a truncated form of desmoplakin in the proband's skin biopsy (Figure 1).
Family StudyThere was no family history worthy of mention. The cardiological protocol performed on the parents and one sister of the proband was normal (Table 1). However, his other sister had an ALVC phenotype (II:2) with ECG, CMR and MSCT results similar to those of the proband (Figure 2, Figure 3). An electrophysiological study in the RV revealed the presence of fragmented potentials and SMVT (with left bundle-branch and inferior axis block), which was readily induced. Her skin desmoplakin expression pattern coincided with that of the proband (Figure 1). Following these results, she was fitted with a defibrillator. The Q1866X desmoplakin mutation, which was absent in the mother of the proband (I:2), was found in the sister with an ALVC phenotype (II:2), and in 2 genetic carriers with a normal cardiological profile: another sister (II:3) and the father (I:1). When genetic results were included, the 2 patients who were affected (II:1 and II:2) met the new ACM criteria.8
Table 1. Results of the Family Study
| Patient | Sex / age | History | ECG | Echocardiogram | Laboratory Tests | ET | 24-h Holter ECG | CMR | MDCT | EMB | EPS | Genetics | DSP Expression |
| I:1 | M/77 | - | SR, 1° AVB, anterior hemiblock | Normal | Normal | No negative submaximum | SR, 0 VEs | Normal | _ | _ | _ | DSP Q1866X in heterozygotes | _ |
| I:2 | F/75 | NYHA II/IV Dyspnea | SR, LVH | Mild AI | Normal | Negative submaximum | SR, 0 VEs | Normal | _ | _ | _ | No mutations | - |
| II:1 (proband) | M/37 | Atypical thoracic pain. AICD | SR, low potentials, 1° AVB, delayed ventricular depolarization (inferior and V1-6 leads), T negative (V3-6 leads) | Normal | (transitory CK [128 U/l]), CK-MB mass (5 U/l) and transaminases (AST 129 U/l, ALT 176 U/l and LDH 758 U/l) during admission for SVT | Negative submaximum, 7 VEs (RBBB and LBBB) | _ | Normal thickness and volumes; LVEF 54% and RVEF 42%. Subepicardial LGE on inferior, lateral and anterior LV, accordion sign in the RV. No infiltration of fatty deposits | _ | Interstitial fibrosis with < 30% loss of myocytes | _ | DSP Q1866X in heterozygotes | DSP I, DSP II and truncated DSP band at 160 kDa |
| II:2 | F/41 | Palpitations, vasovagal syncopes, Sjögren syndrome, Raynaud's disease and probable systemic erythematous lupus. AICD | SR, PR minimal, low potentials, precordial transition in V1, delayed ventricular depolarization (inferior leads), flat Ts in V1-3 and negative Ts in V4-6 leads | General HK, LVEF 45% | Normal | No negative submaximum, 1° AVB, 4 VEs (RBBB and LBBB) | SR, 636 VE | Global HK, patchy thinning of the LV; LVEF 42%; ILVTV 54 ml/m2; RVEF 52%. Septal LGE in LV and subepicardial LGE in inferoseptal, inferior and lateral LV. No infiltration of fatty deposits | Myocardium of the wavy LV indicating fibrous/fatty infiltration | _ | AH 110, HV 41. Fragmented potentials in RV, TVM SMVT inducible (LBBB with inferior axis) | DSP Q1866X in heterozygotes | DSP I, DSP II and truncated DSP band at 160 kDa |
| II:3 | F/40 | Vasovagal syncopes | SR | Normal | Normal | Negative submaximum | SR, 64 VEs | Normal | _ | _ | _ | DSP Q1866X in heterozygotes | _ |
AI, aortic insufficiency; AICD, automatic implantable cardiac defibrillator; AVB, atrioventricular block; CMR, cardiovascular magnetic resonance; DSP, desmoplakin; ECG, electrocardiogram; EF, ejection fraction; EMB, endomyocardial biopsy; EPS, electrophysiological study; ET, effort test; F, female; HK, hypokinesia; ILVTV, indexed left ventricular telesystolic volume; LBBB, left bundle branch block; LGE, late gadolinium enhancement; LV, left ventricle; LVH, left ventricular hypetrophy; M, male; MDCT, multi-detector computed tomography; NYHA, New York Heart Association; RBBB, right bundle branch block; RV, right ventricle; SMVT, sustained monomorphic ventricular tachycardia; SR, sinus rhythm; VE, ventricular extrasystole.
This study highlights the importance of multidisciplinary analysis when families suspected of being affected by ACM are evaluated. It also describes a novel mutation in the desmoplakin gene, indicating the molecular mechanism which is most likely to be implicated.
The ECG of LV ACM revealed flattened/inverted (infero-)lateral repolarization.4, 5, 6, 8 Our patients also had low potentials, as well as delayed ventricular depolarization (inferior leads) and atrioventricular conduction. These novel findings, which have not been included in other articles4, 5, 6 or in the new criteria,8 came to light in a case of Carvajal disease9 and could be useful in confirming suspected ALVC. The electrocardiographic localization of these abnormalites coincides with the widespread fibrosis we detected in the inferolateral lead.
In accordance with our first impressions,10 including the genetic diagnosis increased the sensitivity of the ACM criteria; unfortunately, however, the clinical cardiologist still has limited access to such tests. Although desmocollin-2 and desmoglein-2 mutations have been occasionally described, our results reinforce the view that desmoplakin could be the first gene to study in cases of ACM.4, 11 Immunotransference confirmed the prediction of truncation of desmoplakin in the presence of the Q1866X mutation. Thus, the absence of C-terminals in 50% of the desmoplakin in the affected subjects would explain defective binding between desmosomes and intermediate filaments.4
Although the recommendation of a defibrillator as a primary prevention measure in the affected sister (II:2) was controversial, we advised this treatment because of the presence of risk markers, such as family history, LV disease, and the induction of SMVT in the electrophysiological study.12
The fibrous component predominates over fatty infiltration in ALVC,4, 5, 6 with a subepicardial LGE pattern similar to that of various pathologies (myocarditis, infiltrative myocardiopathies, sarcoidosis, Chagas disease, dilated myocardiopathy, dystrophinopathies, etc.).13 The first CMR suggested a diagnosis of myocarditis in our proband and, without the family study, it would not have been correctly classified. We need to remember that cases of myocarditis can trigger episodes of ACM activity and that the absence of typical ACM histological data does not rule out the diagnosis. The wavy contours that we observed in the MDCT images corroborated the CMR findings and could be useful in patients who are fitted with a defibrillator.
To summarize, the diagnosis of ALVC is complex and involves tests which are not always available, and family assessment may be pivotal in the differential diagnosis.
FundingThis study was funded by grants from the Instituto de Salud Carlos III (PI070831, CP0700326, RECAVA RD06/0014/0004), the Sociedad Valenciana de Cardiología, the Heart Rhythm Society and the British Heart Foundation.
Conflicts of interestNone declared.
Acknowledgements
We would like to thank Dr Monserrat Évole, Dr Aitana Braza-Boïls and Dr Antonio Moscardó for their collaboration in the application of the techniques which were used.
Received 27 April 2010
Accepted 12 October 2010
Corresponding author: Departamento de Cardiología, Hospital La Fe, Avda. Campanar 21, 46009 Valencia, Spain. zorio_est@gva.es