Fabry disease is a rare progressive X-linked sphingolipid storage disorder caused by deficiency of lysosomal a-galactosidase A (a-galA) due to mutations in the GLA gene. The disease triggers an intracellular accumulation of globotriaosylceramide in various tissues and results in multiple organ damage. Male patients with the classic form develop early signs and symptoms in childhood or adolescence.1 This classic form typically appears in male carriers of genetic variants that cause a severe decrease (or complete absence) of the a-galA enzymatic activity, as occurs with nonsense and frameshift variants.2 Females are heterozygous for mutations in the GLA gene and show a heterogeneous clinical spectrum that ranges from asymptomatic to a clinical severity equal to that of males.3
We report the case of a male Fabry disease patient who developed a relatively mild phenotype despite carrying a classic nonsense GLA variant.
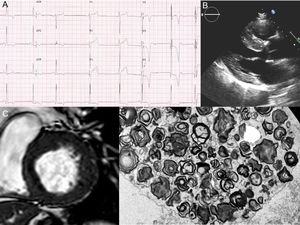
A 58-year-old male patient with hypertension and proteinuria was admitted to hospital due to abdominal pain. On electrocardiography, a short PR interval with left ventricular hypertrophy and subepicardial ischemia was observed (Figure 1A). A coronary angiogram was performed, showing a distal occlusion of the second diagonal branch; symptoms improved with medical treatment. His echocardiogram and cardiac magnetic resonance demonstrated a basal septal hypertrophy of 20 mm (Figure 1B, C). With a diagnosis of hypertrophic cardiomyopathy, the patient was referred to the inherited cardiovascular diseases unit. Fabry disease was suspected due to the presence of hypertrophic cardiomyopathy, proteinuria with mild renal failure (Cr 1.3 mg/dL), and a short PR on electrocardiography. A-galA activity in blood was reduced to 0.7μmol/L/h (2.0-11.7), and renal biopsy showed typical “zebra bodies” on electron microscopy images (Figure 1D). Finally, 17 genes related to hypertrophic cardiomyopathy, including GLA, were sequenced by next-generation sequencing. A previously described truncating mutation in GLA (p.Gln386*/g.10021C>T) associated with classic forms of Fabry disease was identified.4 Therefore, the diagnosis was confirmed and enzyme replacement therapy was initiated.

A: the patient's electrocardiogram showing short PR interval, left ventricular hypertrophy, and subepicardial ischemia. B and C: echocardiogram and cardiac magnetic resonance with left ventricular hypertrophy. D: electron microscopy images of the renal biopsy with the “zebra bodies” (cluster of glycolipid concentric membranous bodies sequestered within lysosome).
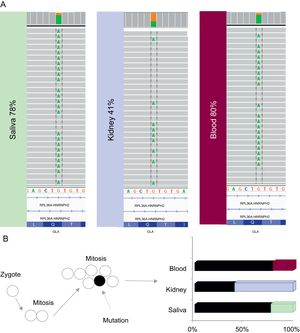
The genetic study of the index case was initially performed on a saliva sample and showed that 78% of the readings had the mutated allele, when 100% is expected in hemizygous carriers; the mutation was confirmed by Sanger sequencing. An additional next-generation sequencing study was carried out in blood and paraffin samples (renal biopsy), showing that 80% and 41% of the readings, respectively, had the mutated allele, confirming the presence of a somatic mosaicism (Figure 2A). However, the Sanger method is imprecise when quantifying the percentage of mosaicism and evaluating the differences between tissues.
 that shows the mutation (discontinued arrow) and another (white) wild-type. Bar diagram showing the proportion readings with the mutated allele.")
A: the readings obtained in the next-generation study of saliva, kidney and blood samples are shown between discontinued black lines. B: two cellular lines in the somatic cells of the patient: one (black) that shows the mutation (discontinued arrow) and another (white) wild-type. Bar diagram showing the proportion readings with the mutated allele.
Both an unaffected daughter and sister of the proband (aged 30 and 55 years) were noncarriers of the variant.
This patient has a phenotype that is milder than that expected for a male carrier of a truncating mutation, in which the involvement of different organs begins at an early age and the phenotype is severe due to the total absence of the enzyme. In this particular case, this can be explained by the presence of 2 cell lines in the somatic cells of the patient: one that shows the mutation and another that is wild-type. Thus, in each tissue, there is a proportion of cells capable of maintaining certain levels of enzyme activity. This is also reflected in the a-galA activity of this patient, which is low but not in the levels expected for a hemizygous null GLA mutation carrier (Figure 2B).
Fabry disease is inherited in an X-linked manner, and therefore an affected male would transmit the pathogenic variant to all of his daughters. In this case, female offspring would inherit the mutation depending on whether the mosaicism is present in the germ cells of the patient or not. In the first case (a germinal mosaicism is present) the risk of inheriting the mutation in females is related to the proportion of mutated sperm.5 Regrettably, this cannot be determined in this patient, because genetic testing was not performed on sperm tissue and the only genotyped daughter is a noncarrier of the variant. We have reported the first case of FD caused by a somatic mosaicism in the GLA gene. The patient exhibited a milder phenotype than that expected for a hemizygous carrier.
This case shows the importance of next-generation sequencing technologies in the genetic testing of patients with suspected Fabry disease, allowing detection of patients in early stages (even those with somatic mosaicism such as this case) who may benefit from enzyme replacement therapy.6
FUNDINGSupported by grant from the Centro de Investigación Biomédica en Red (CIBERCV), “Instituto de Salud Carlos III”, CB16/11/00425 (R. Barriales-Villa and L. Monserrat), “Instituto de Salud Carlos III”, FEDER “Unión Europea, Una forma de hacer Europa”
CONFLICTS OF INTERESTJ.P. Ochoa and J.L. Santomé-Collazo are employees of Health in Code S.L. R. Barriales-Villa have received personal fees from Health in Code S.L. L. Monserrat is a stakeholder and CEO of Health in Code S.L.