
We report the case of a 9-year-old boy, with no personal or family history of cardiovascular disease, who presented with out-of-hospital cardiac arrest while playing soccer at school. After 7minutes of advanced resuscitation maneuvers, the patient recovered spontaneous circulation.
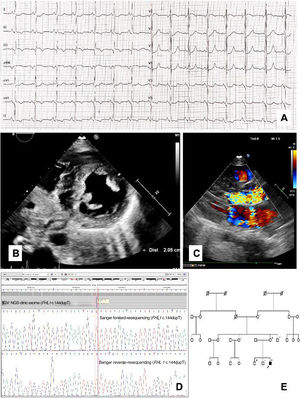
On admission to the pediatric intensive care unit, the patient's electrocardiogram (ECG) showed sinus rhythm, flattened T waves and ST segment depression in inferolateral leads, and pathologic Q waves in aVR (figure 1A). An echocardiogram showed asymmetric left ventricular hypertrophy (LVH) with 21-mm interventricular septum (Z score > +6), preserved systolic function, and systolic anterior movement of the mitral valve, resulting in a gradient through the left ventricular outflow tract (LVOT) up to 90mmHg with posterior moderate mitral regurgitation (figure 1B,C). Neurological examination was normal, with no involvement of limb muscles or joints, and no anomalous CK rise. Twelve days after admission, a subcutaneous implantable cardioverter-defibrillator (ICD) was implanted for secondary prevention. The patient made a successful recovery, started treatment with propranolol, and remained asymptomatic until 1 year later when he had an appropriate ICD shock.

A: Index case electrocardiogram at diagnosis showing abnormal repolarization in inferolateral leads and pathologic Q waves in aVR. B: parasternal short axis 2-dimensional echocardiogram at the level of the papillary muscles showing asymmetric LVH. C: parasternal long axis view of transthoracic color flow Doppler echocardiogram showing left ventricular outflow tract obstruction and moderate posterior mitral valve regurgitation. D: FHL1 c.144dupT variant detection and validation in Integrative Genomics Viewer and Chromas-Sanger Viewer. E: family pedigree, the arrow points to the index case. IGV, integrative genomics viewer; NGS, next-generation sequencing.
A blood sample was collected for clinical exome analysis by next-generation sequencing that included 199 cardiac disease-related genes. A novel nonsense variant in exon 2 of the FHL1 gene (X chromosome) was identified in hemizygosis: NM_001159699: c.144dupT; p. (Asp49 *), considered probably pathogenic (figure 1D). No other relevant variants were found in HCM-associated genes.
The patient's mother and 2 brothers underwent genetic and clinical screening. The mother proved to be a carrier of the variant, with normal phenotype. The siblings did not have an altered echocardiographic phenotype, although the ECG of the eldest brother showed subtle abnormalities such as pathologic Q waves and flattened T waves in lateral leads. The result of the genetic segregation study was negative in the younger brother and the variant was present in the older brother. The maternal grandmother, aunts and uncle were also tested, all of them showing a normal phenotype and none carried the mutation (figure 1E). The proband's deceased maternal grandfather could not be tested. In this scenario, none of the aunts and uncle and neither grandmother are FHL1 c.144dupT carriers and therefore we presume that the variant could be a de novo mutation in the mother.’
We describe the case of a child with hypertrophic cardiomyopathy (HCM) whose first symptom was an episode of resuscitated cardiac death that was positive for a novel likely pathogenic variant in a nonsarcomeric gene with X-linked inheritance pattern. FHL1 has been postulated as a pathogenic gene involved in the etiology of several skeletal myopathies, sometimes in association with dilated, noncompaction or, more commonly, hypertrophic cardiomyopathy.1,2 It is located on chromosome Xq26.3 and encodes for 4.5 LIM domain 1, a LIM protein family member. Each LIM domain contains 2 binding zinc fingers, which are highly preserved. Mutations affecting these regions are usually pathogenic.2,3FHL1 is the main isoform in skeletal and cardiac muscle and plays an important role in sarcomeric protein synthesis, intracellular integrity and signaling, gene transcription, and biomechanical stress sensing.2
Moreover, there is increasing evidence that certain mutations in FHL1 could also explain the pathogenesis of rare cases of isolated HCM that cannot be explained by traditional sarcomeric mutations. FHL1 is the only member of the LIM protein family with different splicing variants, and its mutations lead to harmful peptides resulting in cardiac hypertrophy, diastolic dysfunction, or myocardial contractility impairment.1,4
Table 1 summarizes the few previously published cases and families with HCM without myopathic involvement that have been related to variants in FHL1 (heterozygous or hemizygous). The LVH pattern is heterogeneous, and there are some reported cases of “spongy” anatomy without meeting LV noncompaction criteria.4,5 Male carriers of truncating variants tend to have a very aggressive and early clinical course both in terms of advanced heart failure and malignant ventricular arrhythmias. In contrast, the heterozygous women described to date have milder cardiac involvement.1
Previously described cases of isolated HCM due to mutations in FHL1.
| Sex and age at diagnosis | FHL1 variant | Cardiac phenotype | Cardiovascular events | Reference |
|---|---|---|---|---|
| Male, 16 y | c.134delA, p.Ser45fs.Nonsense. | HCM, septal predominance. LVOT obstruction. IVS 20mm. ECG disturbances. | Friedrich et al. Hum Mol Genet. 2012.2 | |
| Female, 43 y | c.459C>A, p.Cys153X.Nonsense. | HCM. IVS 17mm. ECG disturbances. | Friedrich et al. Hum Mol Genet. 2012.2 | |
| Male, 18 y | c.599_6000insT, p.F2000fs32X.Nonsense. | Isolated HCM, restrictive. IVS 24mm. Moderate MR. ECG disturbances. | Heart transplant at 31 y (end stage HF). | Hartmannova et al. Circ Cardiovasc Genet. 2013.1 |
| Male, 55 y | c.599_6000insT, p.F2000fs32X.Nonsense. | Isolated HCM. IVS 20mm. Moderate MR. ECG disturbances. | Heart transplant at 59 y (end stage HF). | Hartmannova et al. Circ Cardiovasc Genet. 2013.1 |
| Male, 38 y | c.468_469insC.NM_001159700_1.Nonsense. | HCM, apical and midventricular predominance. IVS 17mm. Biatrial enlargement. Diastolic dysfunction. LVEF 56%. MRI late gadolinium enhancement. | Atrial fibrillation.Death at 61 y (pulmonary fibrosis, pulmonary and multiorgan failure). | Gallego-Delgado et al. Int J Cardiol. 2015.3 |
| Female, 32 y | c.468_469insC.NM_001159700_1.Nonsense. | HCM, apical and midventricular predominance. IVS 21mm. Diastolic dysfunction. LVEF 73%. MRI late gadolinium enhancement. | Atrial fibrillation.Stroke at 46 y. | Gallego-Delgado et al. Int J Cardiol. 2015.3 |
| Male, 8 y | c.468_469insC.NM_001159700_1.Nonsense. | Asymmetric HCM. IVS 27mm. LVOT obstruction. Restrictive LV. LVEF 23%. | Aborted SCD (VT) at 8 y. Heart transplant at 17 y. | Gallego-Delgado et al. Int J Cardiol. 2015.3 |
| Male, 7 y | c.468_469insC.NM_001159700_1.Nonsense. | Obstructive asymmetric HCM. IVS 19mm. Moderate MR. | Primary prevention ICD at 11 y. Appropriate shock at 17 y.SCD (VT, with lower HR than ICD threshold) at 17 y. | Gallego-Delgado et al. Int J Cardiol. 2015.3 |
| Male, 19 y | c.267C>A, p.Cys89Ter.Nonsense. | HCM with extensive interstitial fibrosis at autopsy. IVS 20mm. | Sudden cardiac arrest. | Gaertner-Rommel et al. Mol Genet Genomic Med. 2019.5 |
ECG, electrocardiogram; HCM, hypertrophic cardiomyopathy; HF, heart failure; ICD, implantable cardioverter-defibrillator; IVS, interventricular septum; LV, left ventricle; LVEF, left ventricular ejection fraction; LVOT, left ventricular outflow tract; MR, mitral regurgitation; MRI, magnetic resonance imaging; SCD, sudden cardiac death; VT, ventricular tachycardia.
The FHL1 c.144dupT variant has not been previously described in patients and is not present in population databases. Like other pathogenic/likely pathogenic variants in FHL1, the mutation leads to a truncated protein. In this particular case, the variant is predicted to lead to a shortened protein (49 amino acids instead of 296 amino acids, losing all functional elements). We believe it to be likely pathogenic according to the modified American College of Medical Genetics and Genomics (ACMG) criteria.6 Another patient previously reported in 2012 with isolated HCM showed a mutation with a very close genomic location, c.134delA (p.Ser45fs).2
The index patient had a cardiac arrest at the age of 9 years. To the best of our knowledge, there is only one report of a similar case.3 The authors describe a family with a novel frameshift variant in FHL1 and isolated HCM in which an 8-year-old boy was diagnosed after collapsing due to ventricular tachycardia. Regarding the rest of the family, the mother had a normal phenotype at the first evaluation. This finding is in keeping with previous cases reported of females with mild phenotype and similar to other diseases with an X-linked inheritance pattern. The oldest brother carried the variant although he did not have a clear phenotype when first screened. However, incomplete penetrance and variable expressivity are common in HCM as well as in other cardiomyopathies. Of note, the patient is still very young and therefore a close follow up is warranted.
Sudden cardiac death (SCD) as a first symptom in HCM is very unusual in the pediatric population but potentially devastating. Currently, a great deal of research is attempting to determine the main risk factors that should be considered to avoid malignant ventricular arrhythmias in children with HCM. Genetic testing is very useful to confirm diagnosis, for family screening, and genetic counseling. However, with few exceptions and cases of double mutation, most HCM causal variants do not currently provide prognostic information on arrhythmic risk. Current available genetic testing with large gene panels or exomes may provide more information on SCD risk stratification, although the results should be interpreted carefully.
Nevertheless, regarding FHL1-related HCM, we believe that published evidence and the case we report here show that young male patients with isolated HCM and truncating variants in FHL1 seem to present with an aggressive course. Hence, male FHL1 mutation carriers with HCM could be considered for early ICD implantation.
The authors accept full responsibility for the content of the manuscript as defined by the International Committee of Medical Journal Editors. The research reported was carried out in accordance with internationally accepted recommendations for clinical investigation (Declaration of Helsinki of the World Medical Association, revised October 2013). The authors confirm they obtained written informed consent from the patient's mother for publication in Rev Esp Cardiol and dissemination in print and electronic form.
FUNDINGThe authors received no financial support.
AUTHORS’ CONTRIBUTIONSM. López Blázquez: conception and design of the work, acquisition of data for the work, drafting the work, final approval of the version to be published, ensuring the accuracy and integrity of the work. A.I. Fernández Ávila: analysis and interpretation of data for the work, final approval of the version to be published. R. Álvarez García-Rovés: acquisition of data for the work. M. Centeno Jiménez: acquisition of data for the work. C. Gómez González: revising the content of the work. M.Á. Espinosa Castro: conception and design of the work, revising the content of the work, final approval of the version to be published.
CONFLICTS OF INTERESTWe declare no potential conflict of interest.