To the Editor,
Long QT syndrome is characterised by a prolongation of the QT interval on the electrocardiogram (ECG), which predisposes to the development of torsade de pointes ventricular arrhythmias. QT prolongation is defined as a corrected QT of >450ms in men and of 470ms in women.1 There are two main groups: congenital long QT syndromes, associated with mutations in certain genes, and the acquired variant, associated with environmental factors.2
The main cause of acquired long QT is pharmacological, and there is a great variety of drugs associated with prolonging the QT interval.3 Other causes include electrolyte disorders (mainly hypokalaemia, hypomagnesaemia and hypocalcaemia, toxic substances like organophosphates, liquid protein intake, endocrine disorders like hypothyroidism4 or phaeochromocytoma, starvation, anorexia nervosa or bradyarrhythmias.
We present the case of a white 50-year-old man who was admitted to hospital because he suffered a syncopal episode while he was driving, causing an accident. The only interesting aspect of his medical history was that he was diagnosed with arterial hypertension 3 months before. He started treatment with amlodipine 5mg, valsartan 160mg and hydrochlorothiazide 12.5mg, with adequate control. Since then he said that he continually felt thirsty, had polyuria and polydipsia, without having previously presented with dizziness or syncope.
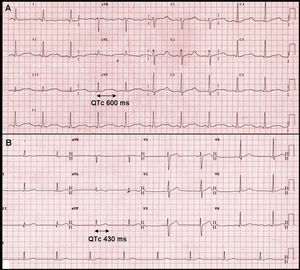
When he arrived at the emergency department he was conscious and aware of his surroundings, with a Glasgow coma score of 15, blood pressure of 150/90mm Hg, and heart beat of 59bpm. Cardiorespiratory and neurological examination was normal. The ECG showed a sinus rhythm with a very significant QT interval prolongation; corrected QT using Bazzet's formula was 600ms (Figure 1A). The analytical tests showed an important electrolyte alteration with hypokalaemia (2.1mEq/l) and hypernatraemia (150mEq/l). Magnesium was normal and blood gas showed mild alkalosis (pH 7.5). In light of these findings, the patient was admitted to the intermediate cardiology care unit. It was initially suspected that a disorder of the renin-angiotensin-aldosterone axis had caused the symptoms.

Figure 1. A, electrocardiogram showing an intense hypokalemia with prolonged QT interval. B, electrocardiogram after resolving hypokalemia with normal QT interval.
High doses of potassium supplements were needed to correct the hypokalaemia, with gradual normalisation of the QT interval duration until normal values were restored (Figure 1B). To control arterial hypertension, calcium antagonists and alpha blockers were initially used, which obtained a suboptimal control. Angiotensin-converting enzyme inhibitors and anti-aldosterone agents had to be added once the hormone studies had been performed to achieve adequate control.
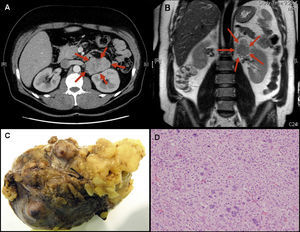
A transthoracic echocardiogram was performed, which ruled out structural heart disease. In view of an aetiological diagnosis, the adrenal function was examined, which showed high plasma aldosterone concentrations and absence of plasma renin activity. A thoracoabdominal computerised tomography scan was also performed showing a lobulated homogeneous tumour of 6.8cmx6.2cmx4cm dependent on the left adrenal gland (Figure 2A) which did not affect other organs. Diagnosis was therefore primary aldosteronism caused by the left adrenal mass.

Figure 2. Computerized tomography image (A) and magnetic resonance (B) of left adrenal mass. Macroscopic (C) and microscopic (D) image of tumor. L, left; R, right.
In the follow-up he initially presented with abundant ventricular extrasystoles of different morphologies (couplets and triplets) but with no sustained arrhythmic events that were gradually remitting with QT normalisation. He was discharged in a stable situation, with potassium supplements and antihypertensive treatment, waiting for the adrenal mass to be surgically removed.
The patient continued to be asymptomatic, and a strict analytical control was performed to keep ion levels within the normal range. Pre-operative magnetic resonance imaging was performed: the tumour heterogeneously enhanced after gadolinium injection and small enlarged lymph nodes were found in the retroperitoneal space (Figure 2B). As planned, the patient underwent surgery to remove the left adrenal tumour and associated lymphadenectomy. The pathological anatomy examination (Figure 2C and D) characterised the mass as an adrenocortical carcinoma with no signs of metastasis in lymph nodes. A variable architecture was observed in the microscopic examination. It was formed by clear cytoplasm cells with important nuclear pleomorphism, areas of frequent necrosis and mitosis, some being atypical. Invasion of the capsule was observed, but no venous invasion. Following the intervention, the electrolyte and blood pressure readings normalised, meaning that potassium supplements and antihypertensive treatment could be withdrawn.
Our patient presented an acquired QT interval prolongation related to an intense hypokalaemia. Once the electrolyte disorder was corrected, normal values were restored. Within a clinical context, we assumed that the patient's syncope was secondary to a torsade de pointes arrhythmic event related to intense hypokalaemia. As with etiologies such as this one, the initial suspicion was confirmed and the diagnosis reached was primary aldosteronism secondary to adrenocortical carcinoma.
The adrenal carcinoma is a very aggressive and rare condition, with an incidence of1-2 cases/million habitants/year.5 Approximately 60% of these are functioning, and the most frequent disorder is Cushing's syndrome. Adrenal carcinoma as a cause of primary hyperaldosteronism is extremely rare, with an incidence of 1%-3% of all cases.6 Surgical resection is the only treatment that has proven effective for tumours and local lymphadenopathies. If this condition is presented as a primary aldosteronism, it may allow for an earlier diagnosis and intervention on the neoplasia.
.
Corresponding author: lgarbue@googlemail.com