
El tronco arterioso común es una cardiopatía congénita troncoconal que representa hasta el 4% de todas las cardiopatías congénitas y se caracteriza por un solo tronco arterial del cual emergen las arterias coronarias, pulmonar y sistémica. La clasificación más utilizada es la de Collet y Edwards, basada en el sitio de nacimiento de las arterias pulmonares. Sin tratamiento quirúrgico, el 80% de los pacientes mueren tempranamente, con supervivencia excepcional en la adolescencia y la edad adulta, y casi todos los sobrevivientes sufren enfermedad obstructiva vascular grave o síndrome de Eisenmenger1,2.
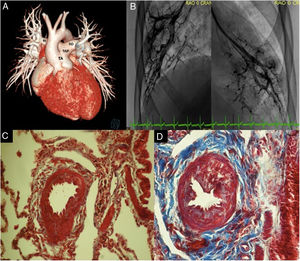
Previa autorización del comité de ética y con el consentimiento de los padres del paciente, se presenta el caso de un varón de 15 años —diagnosticado a los 2 meses de vida de tronco arterioso común tipo I que, por decisión de los padres, se dejó a su libre evolución sin tratamiento quirúrgico— al que se revalúa por disnea. Ingresó con oximetría de pulso del 85% y soplo sistólico paraesternal izquierdo. El ecocardiograma trastorácico confirmó el diagnóstico previo, con válvula troncal trivalva con insuficiencia moderada y comunicación interventricular de 20×26 mm. Se complementó con tomografía computarizada, que mostró el origen anómalo de la coronaria izquierda (figura 1A). El cateterismo evidenció una presión pulmonar sistémica, con presión telediastólica de los ventrículos izquierdo y derecho de 15 mmHg. Al calcular por el método de Fick con consumo de oxígeno presunto de 132 ml/min/m2, el Qp/Qs fue 4,7, con resistencia vascular pulmonar indexada de 5,45 UW/m2 e índice PVR/SVR=0,19; al administrar oxígeno, la relación QP/QS se incrementó a 5,08 y la resistencia vascular indexada disminuyó a 4,49 UW/m2 (tabla 1); además, la mancha capilar derecha fue homogénea (figura 1B). Además se informó cariotipo normal y estudio de hibridación in situ fluorescente (FISH).
; grado I de Heath y Edwards. D: biopsia pulmonar izquierda, con engrosamiento de la pared vascular por músculo liso (rojo) y proliferación de la íntima, con tejido fibroso (azul); grado II de Heath y Edwards. RD: rama pulmonar derecha; RI: rama pulmonar izquierda; TA: tronco arterioso común, TAP: tronco arterial pulmonar. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
A: reconstrucción 3D. Tronco arterioso común tipo I. B: angiografías pulmonares magnificadas en cuña con mancha capilar derecha e izquierda. C: tinción de Masson. Biopsia pulmonar derecha, con proliferación de la pared por hiperplasia de musculo liso (rojo); grado I de Heath y Edwards. D: biopsia pulmonar izquierda, con engrosamiento de la pared vascular por músculo liso (rojo) y proliferación de la íntima, con tejido fibroso (azul); grado II de Heath y Edwards. RD: rama pulmonar derecha; RI: rama pulmonar izquierda; TA: tronco arterioso común, TAP: tronco arterial pulmonar. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
Cateterismo cardiaco con FiO2 al 21% prequirúrgico y 23 meses después de la cirugía
| SaO2 prequirúrgica y posquirúrgica, % | Presión sistólica prequirúrgica y posquirúrgica, mmHg | Presión diastólica prequirúrgica y posquirúrgica, mmHg | Presión diastólica final prequirúrgica y posquirúrgica, mmHg | Presión media prequirúrgica y posquirúrgica, mmHg | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| VCS | 54 | 70 | ||||||||
| VCI | 67 | 81 | ||||||||
| VD | 85 | 61 | 15 | 8 | ||||||
| TAP | 90 | 78 | 85 | 52 | 35 | 9 | 60 | 28 | ||
| VI | 95 | 95 | 99 | 100 | 15 | 11 | ||||
| AoD | 95 | 95 | 99 | 100 | 44 | 53 | 63 | 85 | ||
| SVM | 57,25 | 72,75 | ||||||||
AoD: aorta descendente; SaO2: saturación de oxígeno; SVM: saturación venosa mixta; TAP: tronco arteria pulmonar; VCI: vena cava inferior; VCS: vena cava superior; VD: ventrículo derecho; VI: ventrículo izquierdo.
Se decidió la corrección quirúrgica con conexión del ventrículo pulmonar con tubo de woven-dacron de 22 mm, colocación de prótesis valvular aórtica bovina de 21 mm en posición pulmonar, cierre de la comunicación interventricular con parche de pericardio bovino y plastia de la válvula aórtica. La biopsia pulmonar derecha e izquierda informó muscularización de la capa media de las arterias intralobular y centrolobular, sin reacción de la íntima, vasos pequeños con trombos de fibrina y macrófagos alveolares con hemosiderina (figura 1C,D).
Cursó con una adecuada evolución, en clase funcional NYHA I y con un cateterismo de seguimiento a los 23 meses que registró presión pulmonar media de 28 mmHg y resistencia vascular pulmonar indexada de 4,92 UW/m2 con índice PVR/SVR=0,19.
Excepcionalmente, hay pacientes que, dejados a su historia natural, sobreviven a la adolescencia y la edad adulta; casi todos sufren una enfermedad obstructiva vascular pulmonar grave o síndrome de Eisenmenger2. Los mecanismos fisiopatológicos que determinan la naturaleza reversible o irreversible de la hipertensión pulmonar en las cardiopatías congénitas siguen sin ser claras. El flujo sanguíneo y la presión son factores desencadenantes esenciales para la remodelación vascular pulmonar en las cardiopatías congénitas; así, a mayores flujo y presión, se perturba el flujo sanguíneo en todo el lecho arterial pulmonar, lo que provoca inflamación y proliferación3.
Existen factores de susceptibilidad genética que podrían predisponer a la remodelación vascular pulmonar o acelerarla; los más destacados son el receptor de la proteína morfogenética ósea tipo 2 (BMPR2) y el factor de transcripción Sox174. Roberts et al.5 identificaron mutaciones en el 6% de los pacientes con hipertensión pulmonar asociada con cardiopatías congénitas. Por otra parte, Liu et al.6 encontraron una diferencia significativa de la mutación de BMPR2 entre los pacientes con cardiopatías congénitas y enfermedad vascular pulmonar y aquellos sin enfermedad.
Nuestro caso tiene una inusual historia natural de la enfermedad. El paciente no contaba con alteraciones anatómicas protectoras (estenosis/hipoplasia de ramas pulmonares) ni bandaje pulmonar; además, ha residido en una ciudad a 2.240 m de altitud, y al no desarrollar precozmente hipertensión pulmonar irreversible a pesar de un cortocircuito importante, podría relacionarse con procesos de adaptación fisiológicos y genéticos descritos en animales que viven a gran altitud. Se desconoce si existen factores genéticos protectores que expliquen por qué algunos pacientes, a pesar de tener cardiopatías congénitas con cortocircuito importante, contraen una hipertensión pulmonar tardía y menos grave.
En conclusión, se debe estudiar integralmente a los pacientes candidatos para corrección quirúrgica sin considerar la edad como criterio para descartar el tratamiento correctivo, y es importante comprender qué mecanismos permiten que algunos pacientes estén protegidos y no sufran enfermedad vascular pulmonar.
FINANCIACIÓNNo se recibió ningún patrocinio para llevar a cabo este artículo.
CONTRIBUCIÓN DE LOS AUTORESConcepto/diseño: J. Calderón-Colmenero, J. A. García-Montes, J. L. Cervantes-Salazar. Redacción: A. Aranda-Frausto, F. Castillo-Castellón, E. Lupinta-Paredes. Revisión crítica/aprobación: J. Calderón-Colmenero, J. A. García-Montes, A. Aranda-Frausto, F. Castillo-Castellón, E. Lupinta-Paredes, J. L. Cervantes-Salazar.
CONFLICTO DE INTERESESLos autores declaran no tener ningún conflicto de intereses.