El síndrome de Brugada (SB) es una canalopatía que se asocia con riesgo de arritmias ventriculares malignas y muerte súbita. El diagnóstico se hace con base en el patrón observado en el electrocardiograma (ECG) y las características clínicas, y el tratamiento fundamental es el implante de un desfibrilador, aunque últimamente se están desarrollando prometedoras técnicas de ablación1. En un alto porcentaje de los casos, el ECG basal es normal o no concluyente, y se requiere una prueba de provocación farmacológica para confirmarlo. Algo similar ocurre con los familiares de los afectados, que pueden heredar la enfermedad. El estudio genético puede identificar la causa, confirmar el diagnóstico y evitar que el paciente y sus familiares se expongan a pruebas o seguimientos innecesarios.
El principal gen implicado es el del canal del sodio SNC5A, pero existen otros genes implicados en menor porcentaje. Un estudio genético se debe considerar completo cuando, además de estudiar estos genes, se descartan variaciones en el número de copias (VNC), como grandes deleciones o duplicaciones, ya que un pequeño porcentaje de casos puede deberse a estas variantes. Se presenta el caso de una familia con SB cuyo estudio genético mediante secuenciación masiva (NGS) permitió identificar la etiología.
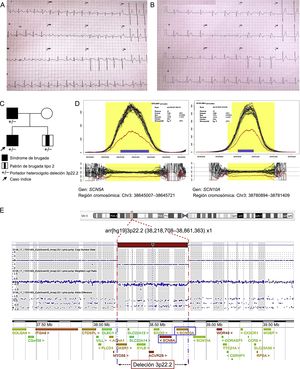
El probando, varón de 13 años, ingresó por palpitaciones tras ejercicio intenso; sin síntomas previos, no estaba en tratamiento farmacológico. Antes había estado en seguimiento por la unidad de arritmias con ECG anual, ya que su padre estaba diagnosticado de SB (presentaba patrón tipo 1 espontáneo en el ECG y un estudio electrofisiológico donde se indujo fibrilación ventricular y se le implantó un desfibrilador). A su ingreso se documentó por ECG un aleteo auricular asociado con un patrón de Brugada de tipo 1 (figura A); el aleteo cedió espontáneamente, y más tarde se registró en el ECG un ritmo nodular con el mismo patrón de tipo 1 (figura B). Se dio el alta al paciente en ritmo sinusal, sin necesidad de medicación. Se le realizó un estudio electrofisiológico ambulatorio, en el cual no se indujeron arritmias ventriculares. Un Holter ECG mostró bradicardia sinusal, pausas sinusales y extrasistolia auricular frecuente, que se interpretó como probable disfunción del nódulo sinusal.
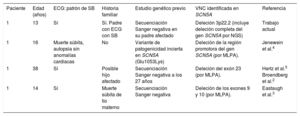
. D: el análisis de coberturas de la NGS muestra una deleción en heterocigosis; eje de abscisas, región genómica; eje de ordenadas, cobertura de la secuencia (número de lecturas); cada línea en negro representa 1 solo caso; la línea azul, la mediana de todos los casos, y la roja, el caso índice. E: deleción 3p22.2 caracterizada mediante SNP-array; la gráfica muestra una disminución de la señal en esta región. NGS: estudio genético mediante secuenciación masiva. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Síndrome de Brugada familiar. A: electrocardiograma del caso índice en aleteo auricular. B: patrón de Brugada tipo 1 posterior. C: pedigrí de la familia; estudio genético familiar (SNP-array). D: el análisis de coberturas de la NGS muestra una deleción en heterocigosis; eje de abscisas, región genómica; eje de ordenadas, cobertura de la secuencia (número de lecturas); cada línea en negro representa 1 solo caso; la línea azul, la mediana de todos los casos, y la roja, el caso índice. E: deleción 3p22.2 caracterizada mediante SNP-array; la gráfica muestra una disminución de la señal en esta región. NGS: estudio genético mediante secuenciación masiva. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
El padre del niño nunca había sufrido choques del dispositivo ni se había observado disfunción sinusal o arritmias auriculares. Se le realizó un estudio genético del gen SCN5A previo, mediante secuenciación Sanger, que fue negativo. El hermano del probando, de 11 años de edad, se encontraba asintomático y presentaba un patrón de Brugada tipo 2 en el ECG basal. En el momento que ingresó el probando, no se había realizado test de provocación a los niños. El padre no tiene hermanos. La madre estaba sana/asintomática y con ECG normal. No se conocen más casos en la familia (figura C).
El estudio genético realizado al niño mediante un panel de NGS identificó una deleción completa de los genes SCN5A y SCN10A (figura D). La confirmación mediante otra técnica molecular más específica para el estudio de VNC (SNP-array) permitió caracterizar la deleción (figura E), que incluía 8 genes. De estos, solo 3 se han asociado con enfermedad: SCN5A y SCN10A (SB), y ACVR2B (defectos cardiacos congénitos complejos). El estudio genético familiar identificó la misma deleción (mediante SNP-array, de manera económica y precisa) en su padre afectado y el hermano asintomático.
Esta deleción no se había descrito previamente, y en la base de datos DECIPHER aparece una deleción similar, pero que involucra solo 3 de estos genes (EXOG, SCN5A y SCN10A), considerada patogénica e identificada en un varón sin fenotipo descrito. Diferentes estudios han descrito grandes deleciones parciales del gen SCN5A en pacientes con SB2–5; en todos los casos se trataba de estudios realizados mediante multiplex ligation-dependent probe amplification (MLPA) tras un estudio genético previo negativo (tabla). Finalmente señalamos la coexistencia del aleteo auricular y el SB en el probando. Fisiopatológicamente, el SCN5A hace un papel importante en el automatismo del nódulo sinusal, y las mutaciones que generan una hipofuncionalidad en este canal se han asociado tanto con disfunción sinusal (con presencia de arritmias auriculares) como con el SB6.
Pacientes con diagnóstico de SB con genético previo negativo o no concluyente, en los que posteriormente se ha identificado una deleción del gen SCN5A
| Paciente | Edad (años) | ECG: patrón de SB | Historia familiar | Estudio genético previo | VNC identificada en SCN5A | Referencia |
|---|---|---|---|---|---|---|
| 1 | 13 | Sí | Sí. Padre con ECG con SB | Secuenciación Sanger negativa en su padre afectado | Deleción 3p22.2 (incluye deleción completa del gen SCN5A por NGS) | Trabajo actual |
| 1 | 16 | Muerte súbita, autopsia sin anomalías cardiacas | No | Variante de patogenicidad incierta en SCN5A (Glu1053Lys) | Deleción de la región promotora del gen SCN5A (por MLPA). | Jenewein et al.4 |
| 1 | 38 | Sí | Posible hijo afectado | Secuenciación Sanger negativa a los 27 años | Deleción del exón 23 (por MLPA). | Hertz et al.5 Broendberg et al.2 |
| 1 | 14 | Sí | Muerte súbita de tío materno | Secuenciación Sanger negativa | Deleción de los exones 9 y 10 (por MLPA). | Eastaugh et al.3 |
ECG: electrocardiograma; MLPA: multiplex ligation-dependent probe amplification; NGS: estudio genético mediante secuenciación masiva; SB: síndrome de Brugada; VNC: variaciones en el número de copias.
En conclusión, esta es la primera descripción de una deleción completa de los genes SCN5A y SCN10A causante de SB familiar. Representa un claro ejemplo de la utilidad de la NGS en el diagnóstico de estas enfermedades. Hoy es posible mediante NGS realizar un estudio genético completo, que permite identificar variantes puntuales y VNC en los genes de interés, lo cual evita los falsos negativos y permite un adecuado cribado familiar. El desarrollo de estas tecnologías está permitiendo identificar nuevos mecanismos moleculares y ampliar el conocimiento sobre la etiología de estas enfermedades.
CONFLICTO DE INTERESESJ.P. Trujillo-Quintero, J.P. Ochoa y D. de Uña pertenecen al Departamento Clínico Health in Code, una empresa con alta experiencia en el diagnóstico genético de enfermedades cardiovasculares.
AGRADECIMIENTOSA la familia, por depositar su confianza en el equipo clínico, y a todo el personal de laboratorio y bioinformático que hace parte fundamental de este equipo de trabajo.