Los tumores cardiacos primarios son raros y su incidencia en las series iniciales oscila entre el 0,001 y el 0,3%1. Los sarcomas, en especial los que se originan en el pericardio, son extremadamente raros. En este artículo se presenta un caso de sarcoma sinovial pericárdico primario y se revisa la literatura existente sobre este tumor con la finalidad de mejorar el conocimiento de esta entidad y su tratamiento.
Un varón de 58 años de edad consultó por dolor pericárdico. Presentaba una sacroiliítis positiva para HLA (locus del antígeno de histocompatibilidad) B27, pero realizaba ejercicio físico sin limitaciones. No tenía ninguna otra enfermedad de interés, abuso de sustancias o antecedentes familiares de cáncer conocidos.
La ecocardiografía transtorácica, la tomografía computarizada multidetectores, la resonancia magnética (RM) y la tomografía por emisión de positrones con fluorodesoxiglucosa-18 (FDG-PET) (figura) mostraron una masa de 93 × 55mm situada en la pared lateral del ventrículo izquierdo, sin extensión extracardiaca. Se trataba de un tumor friable con tejido necrótico, intensamente adherido al corazón, por lo que fue imposible la resección completa. Mediante el análisis histológico se diagnosticó sarcoma sinovial pericárdico. Aplicando un enfoque multidisciplinario, se remitió al paciente a quimioterapia y radioterapia adyuvante, que obtuvo buena respuesta inicial. Sin embargo, 7meses después, una nueva RM mostró recidiva local del tumor. Se aplicó quimioterapia y se realizó una nueva resección quirúrgica. Sin embargo, se ha presentado una nueva recidiva.
, situada en el pericardio posterolateral. B:FDG-PET, imagen en plano transversal; puede observarse un aumento de la captación en la masa pericárdica. FDG-PET:tomografía por emisión de positrones con fluorodesoxiglucosa-18; RM: resonancia magnética.")
Exploraciones de imagen multimodal. A:RM; la secuencia de RM de gradiente-eco muestra una masa bien delimitada (flecha), situada en el pericardio posterolateral. B:FDG-PET, imagen en plano transversal; puede observarse un aumento de la captación en la masa pericárdica. FDG-PET:tomografía por emisión de positrones con fluorodesoxiglucosa-18; RM: resonancia magnética.
Los sarcomas sinoviales son tumores malignos de tejidos blandos, con un patrón de crecimiento agresivo. Aunque aparecen predominantemente en las extremidades, se ha descrito su presencia en casi todos los órganos. Se clasifican en 4subtipos en función de la proporción de células epiteliales y fusiformes: bifásico, monofásico de células fusiformes, monofásico de células epiteliales y no diferenciado. Este paciente presentaba una variante monofásica con predominio de células fusiformes. Los marcadores inmunohistoquímicos, como la vimentina, el antígeno de membrana epitelial y la citoqueratina, son útiles para descartar otros posibles tumores de tejidos blandos2. El diagnóstico definitivo puede realizarse por la existencia de una translocación cromosómica presente en más del 90% de los casos, y que no se ha observado que se asocie con otros sarcomas. Esta translocación afecta a los genes SSX1 o SSX2 del cromosomaX y el gen SYT del cromosoma18, que forman un gen de fusión, el SYT-SSX, lo que da lugar a la producción de proteínas de fusión3,4.
El diagnóstico se establece mediante técnicas de imagen multimodales y examen histológico del tumor. La tomografía computarizada, la RM y la FDG-PET son útiles para determinar la localización del tumor y evaluar la invasión de órganos adyacentes5. También son esenciales para la planificación quirúrgica y el seguimiento posterior al tratamiento.
El tratamiento es difícil y requiere un abordaje multimodal. La cirugía es la piedra angular del tratamiento y puede aportar un tratamiento curativo. Sin embargo, lo más frecuente es que los tumores estén adheridos a órganos vitales adyacentes y no sean resecables. En consecuencia, la radioterapia y la quimioterapia pueden ser útiles para reducir las recaídas. La doxorubicina y la ifosfamida son los fármacos de quimioterapia más eficaces y pueden alcanzar una alta tasa de respuesta6. La causa más frecuente de muerte es la recidiva local, incluso tras una resección macroscópica completa. Al igual que en el presente caso, pocos pacientes se mantienen sin recaídas a los 12meses, por lo cual la mayoría de ellos requieren tratamientos repetidos.
Hasta donde sabemos, en la literatura médica en lengua inglesa son pocos los casos de sarcoma sinovial pericárdico. En una búsqueda en PubMed con el término “pericardial synovial sarcoma”, en agosto de 2016 solo se identificaron 54referencias. Al examinar estos artículos, así como las publicaciones citadas en ellos, se identificaron solo 38casos de sarcoma sinovial originado en el pericardio (tabla). Se revisaron estos casos en cuanto a epidemiología, cuadro clínico, conducta terapéutica y pronóstico de esta neoplasia. Ocurre predominantemente en varones, con mayor incidencia en la cuarta década de la vida. La mayor parte de los sarcomas eran de tipo monofásico (células fusiformes) y se identificó la translocación citogenética. Como en el presente caso, hubo predominio de inmunohistoquímica positiva para vimentina, antígeno de membrana epitelial y citoqueratina. Todos los casos se trataron con extirpación quirúrgica si era posible, y se aplicó un tratamiento neoadyuvante (quimioterapia y radioterapia) para reducir las tasas de recidiva local, de manera similar al tratamiento del caso presentado aquí. Se realizó una extirpación completa o casi completa del tumor en una minoría de los casos publicados, de manera que la mayoría de los pacientes presentaron recaídas locales o un tiempo de supervivencia corto.
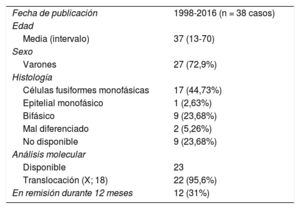
Sarcomas sinoviales pericárdicos publicados
| Fecha de publicación | 1998-2016 (n = 38 casos) |
| Edad | |
| Media (intervalo) | 37 (13-70) |
| Sexo | |
| Varones | 27 (72,9%) |
| Histología | |
| Células fusiformes monofásicas | 17 (44,73%) |
| Epitelial monofásico | 1 (2,63%) |
| Bifásico | 9 (23,68%) |
| Mal diferenciado | 2 (5,26%) |
| No disponible | 9 (23,68%) |
| Análisis molecular | |
| Disponible | 23 |
| Translocación (X; 18) | 22 (95,6%) |
| En remisión durante 12 meses | 12 (31%) |
En conclusión, en esta publicación se describe un caso de sarcoma sinovial de origen pericárdico y una revisión actualizada de la literatura. Este sarcoma tiene mal pronóstico y son cruciales tanto el abordaje diagnóstico sistemático como el tratamiento multidisciplinario. El análisis de las características histológicas del tumor, el curso clínico del paciente y la respuesta al tratamiento permitirán establecer estrategias terapéuticas óptimas para el tratamiento de estos tumores extremadamente raros.