
Palabras clave
INTRODUCCIÓN
La utilización de la resonancia magnética (RM) como técnica de imagen inocua y precisa ha aumentado en gran medida en los últimos años1. Cada día hay más pacientes portadores de dispositivos de estimulación cardiaca (marcapasos [MP], desfibrilador automático implantable [DAI]) para los que es frecuente la solicitud de RM. Se estima que, tras implantarse el dispositivo, cada paciente tiene una probabilidad entre el 50 y el 75% de precisar una RM durante su vida2.
Los primeros datos obtenidos en este campo, hacia los años ochenta, fueron desalentadores. Se detallaron graves complicaciones tanto eléctricas como clínicas, e incluso se describieron casos de muerte3,4. Se estableció así la contraindicación actual en este contexto.
Los dispositivos actuales son de menor tamaño e incluyen materiales con menor probabilidad de interferencia electromagnética. En los últimos años se han realizado pequeños estudios de seguridad5-9 que, si bien no son suficientes para levantar la contraindicación, parecen indicar que no se producen complicaciones importantes siempre que se mantengan ciertas condiciones de seguridad.
Planteamos un estudio prospectivo para valorar los riesgos que supone la RM en estos pacientes.
MÉTODOS
Durante 14 meses (octubre de 2007 a diciembre de 2008) se incluyó a 29 pacientes portadores de dispositivos de estimulación cardiaca a los que se indicó una RM como única prueba diagnóstica posible. Se informó a todos ellos sobre las posibles consecuencias derivadas de la exploración y todos dieron su consentimiento.
Se realizaron 33 exploraciones (5 DAI y 28 MP), 4 de ellas en pacientes dependientes de MP, entendiendo como tal ausencia de ritmo ventricular propio estable por encima de 40 lat/min.
Antes y después de la exploración, se procedió a la interrogación completa de los dispositivos registrando marca, modelo, modo de estimulación, frecuencia de seguimiento, estado de la batería (impedancia y/o voltaje), umbrales de estimulación y detección e impedancia de estimulación. Se analizaron 58 sondas (33 ventriculares y 25 auriculares). El umbral de estimulación se calculó en todos los casos variando únicamente la amplitud del impulso, por lo que se expresa en voltios. Ningún paciente era portador de electrodos abandonados.
Siguiendo las recomendaciones vigentes, en los pacientes dependientes de MP se cambió el modo de estimulación a V00 o D00. En los pacientes portadores de DAI, se desactivaron las terapias antitaquicárdicas. Se programó la detección en bipolar dejando la salida a doble voltaje del umbral de estimulación y la detección a mitad de la amplitud de la onda detectada cuando así lo permitiera el dispositivo.
Todos los estudios se realizaron en equipo de RM de 1,5 T (Siemens Magneton-Avanto®), con un límite de SAR < 4 W/kg en toda la extensión corporal.
Durante la exploración se mantuvo monitorización electrocardiográfica y pulsioximétrica y contacto verbal con el paciente. Como medida de seguridad se dispuso de material de reanimación cardiopulmonar avanzada en la antesala de la RM, con desfibrilador eléctrico externo y el programador de cada dispositivo.
Tras la RM, se aplicó un cuestionario sistemático con parámetros como dolor, sensación de movimiento del dispositivo, calor o molestia. También se registró la duración de la exploración y se preguntó al radiólogo responsable si se había producido alguna deficiencia en la imagen o limitación en la técnica.
La distribución en cuanto a modo de estimulación, marca de los dispositivos y tipo de RM se muestra en la tabla 1.

Los parámetros cuantitativos se analizaron mediante prueba de la t de Student para muestras apareadas. En las series con n < 30 (impedancia y voltaje de batería, impedancia auricular, detección auricular y ventricular), se utilizó la prueba de la U de Mann-Whitney.
Para el análisis de los cambios en el umbral de estimulación, se definió como variable dicotómica la existencia de cualquier variación de dicho umbral. Posteriormente, mediante test de la χ2, se analizó la posible relación con respecto a cámara estimulada (aurícula, ventrículo) y localización infradiafragmática o supradiafragmática de la exploración.
Los datos cualitativos se expresaron en porcentaje y los cuantitativos en media ± desviación típica. Se consideró significación estadística cuando p < 0,05.
RESULTADOS
No se registró ninguna complicación clínica. Ningún paciente refirió dolor, calor, molestia o sensación de movimiento del dispositivo durante la exploración. En todos los casos se completó el estudio sin limitaciones técnicas; se registró un caso de artefacto en las imágenes obtenidas, durante la realización de una RM cardiaca en un MP modelo Vitality 2DR® (Guidant).
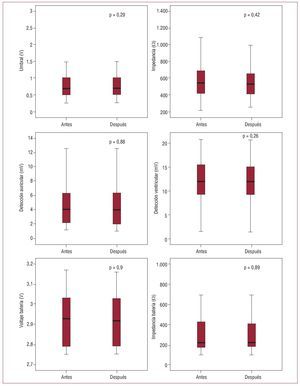
Ninguno de los parámetros eléctricos analizados cambió de forma significativa tras la realización de la RM (tabla 2).

En las 58 sondas expuestas se midió la impedancia de estimulación; no se pudo medir el umbral de estimulación en tres sondas auriculares porque el paciente se encontraba en fibrilación auricular. En 11 casos (20%) se observó una ligera variación en el umbral de estimulación (en 3 de ellos se objetivó aumento de umbral, y en el resto se redujo ligeramente); el máximo cambio registrado fue una disminución de 0,5 V en una sonda ventricular.
De las 11 sondas que sufrieron algún cambio en su umbral de estimulación, 5 fueron ventriculares y 6, auriculares, sin significación estadística en esta relación (p = 0,271). De las 11 exploraciones en las que se registró algún cambio de umbral, 7 fueron infra-diafragmáticas y 4, supradiafragmáticas (p = 0,023).
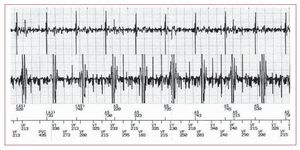
Se registraron dos fallos de detección, uno auricular y otro ventricular (figs. 1 y 2). En el primer caso se registró durante una RM cerebral a un paciente portador de un MP DDD EnPulse E2DR01® (Medtronic®) y el segundo durante una cardiorresonancia a un paciente portador de DAI (Guidant Vitality-2DR®). En ambos casos los pulsos de radiofrecuencia liberados durante la exploración se registraron como ruido en los electrogramas auricular y ventricular respectivamente. Esa señal de ruido se interpretó como fibrilación auricular en el primer caso —el cambio de modo se activó automáticamente— y como fibrilación ventricular en el segundo. Siguiendo el protocolo, previamente se desactivaron las terapias antitaquicárdicas, por lo que ningún caso tuvo relevancia clínica.

Fig. 1. Registro de electrograma auricular y ventricular durante la realización de resonancia magnética a un paciente portador de desfibrilador automático implantable. Se observa señal de ruido en ambos canales, lo que se interpreta como fibrilación ventricular (VF), mientras el paciente conservaba ritmo sinusal estable. El dispositivo no aplicó ninguna terapia, ya que éstas habían sido desactivadas previamente.

Fig. 2. Registro de electrograma de un paciente portador de marcapasos bicameral durante la realización de resonancia magnética. Se observa señal de ruido en canal auricular, que se interpreta como fibrilación auricular y activa el cambio de modo automático (MS). Durante la exploración se objetivó en todo momento ritmo sinusal estable en monitor electrocardiográfico.
Otro evento destacable fue la respuesta de seguridad que presentó uno de los MP analizados durante una colangiorresonancia; era un Clarity DDDR® (Vitatron®) que durante la exploración comenzó con estimulación en modo V00 a frecuencia magnética y salida máxima. Tras la RM, se pudo reprogramar sin problemas a su modo de estimulación previo.
Finalmente, tras dos de las exploraciones, fue preciso reiniciar el programador por incapacidad temporal para establecer conexión telemétrica con el dispositivo de estimulación cardiaca. Tras establecer la conexión, se confirmó ausencia de cambios con respecto a los parámetros previos, y la interrogación tras la RM se llevó a cabo sin ninguna complicación.
DISCUSIÓN
Con esta comunicación intentamos realizar una aproximación científica para determinar con mayor exactitud el riesgo que supone la RM para los pacientes portadores de MP o DAI. Presentamos un estudio prospectivo de 33 casos, todos ellos recogidos entre octubre de 2007 y diciembre de 2008, por lo que tanto las características de los dispositivos de estimulación como las de la RM son similares a las encontradas en la práctica clínica habitual, aunque no deben extrapolarse los resultados obtenidos a otros dispositivos de diferentes marca o modelo.
Los dispositivos estudiados tuvieron una respuesta segura ante la exposición a las interferencias electromagnéticas producidas por la RM. No se registró ningún evento clínico ni cambio significativo en los parámetros eléctricos analizados (fig. 3). Tampoco se produjeron alteraciones irreversibles en el funcionamiento de los dispositivos. Los eventos registrados fueron transitorios tras la reprogramación y no supusieron ninguna limitación en el funcionamiento de los dispositivos ni en la realización de la técnica.

Fig. 3. Representación gráfica comparativa de parámetros eléctricos determinados antes y después de una resonancia magnética. No se observaron cambios estadísticamente significativos en ninguno de ellos.
Debe interpretarse con cautela la relación estadística que indicaría que es más probable que se produzcan cambios de umbral de estimulación al realizar RM infradiafragmática debido al escaso número de cambios de umbral detectados (11 de 50 sondas). Asimismo el reducido número de exploraciones supone una limitación para describir con precisión pequeños cambios en los parámetros analizados, así como la existencia de problemas poco frecuentes.
Los resultados obtenidos son acordes con los de las series publicadas recientemente10,11, y nuestra conclusión está en línea con las recomendaciones generales existentes en la actualidad12. A falta de estudios prospectivos de mayor calado, la realización de RM en estos pacientes debe plantearse de forma individualizada. Los datos actuales indican que, con control cardiológico y manteniendo unas precauciones básicas de seguridad, se podría realizar RM en pacientes portadores de MP y DAI que así lo precisen por razones clínicas, si bien debe destacarse que actualmente no se puede garantizar la absoluta seguridad de una RM en estos pacientes.
Full English text available from: www.revespcardiol.org
Correspondencia: Dr. F. Buendía Fuentes.
Calderón de la Barca, 9, pta. 18. 46010 Valencia. España.
Correo electrónico: franbuendia@gmail.com
Recibido el 30 de abril de 2009.
Aceptado para su publicación el 21 de julio de 2009.