
Los polimorfismos de nucleótido único situados en un lugar de unión de microácidos ribonucleicos (miARN) pueden tener diferentes efectos en la expresión génica, y ello puede influir en el riesgo de enfermedad. Este estudio tiene como objetivo evaluar la asociación existente entre los polimorfismos de nucleótido único y los haplotipos presentes en la región 3’UTR del gen GATA4 y el riesgo de cardiopatía congénita.
MétodosSe utilizaron algoritmos de bioinformática para analizar los polimorfismos de nucleótido único en los presuntos lugares de unión de miARN en la región 3’UTR del gen GATA4 y para calcular la diferencia de energía de hibridación libre (ΔFE, kcal/mol) para cada alelo de tipo natural (wild-type) en comparación con cada variante alélica.
ResultadosFormaron la población de estudio 146 pacientes caucásicos (73 varones; edad, 6,68 ± 7,79 años) y 265 recién nacidos sanos (147 varones). Se consideró que la suma de todos los ΔFE predecía la importancia biológica de los polimorfismos de nucleótido único al unirse a más miARN. A continuación se determinó el genotipo de los 4 polimorfismos (+1158 C > T,+1256 A > T,+1355 G > A, +1521 C > G) que tenían el valor predicho de ΔFE total más alto (9,91, 14,85, 11,03 y 21,66kcal/mol respectivamente) en un estudio de casos y controles (146 pacientes y 250 controles). Al aplicar una corrección por multiplicidad de pruebas, tan solo el alelo +1158 T mostró una diferencia significativa entre los pacientes y los controles. El análisis de los haplotipos puso de manifiesto que el haplotipo T-T-G-C (más infrecuente en los pacientes con cardiopatías congénitas que en los controles) se asociaba a una disminución del riesgo significativa (p = 0,03), mientras que el haplotipo muy infrecuente C-A-A-C, que se daba de manera muy poco común en los controles (0,3%) en comparación con los pacientes con la enfermedad (2,4%), se asociaba a un aumento de 4 veces en el riesgo de enfermedad (p = 0,04).
ConclusionesLas variantes frecuentes de la región 3’UTR del gen GATA4 interaccionan de manera conjunta y con ello afectan a la susceptibilidad a la cardiopatía congénita, probablemente mediante la alteración de la regulación postranscripcional de los miARN.
Palabras clave
Las cardiopatías congénitas (CC) son los defectos neonatales de mayor prevalencia (entre 75 y 90 de cada 10.000 nacidos vivos en los últimos 20 años) y la principal causa de muerte por malformaciones congénitas durante el periodo neonatal y en el primer año de vida1. Las CC comprenden un grupo heterogéneo de defectos cardiacos que surgen durante el desarrollo fetal. Hasta la fecha, los mecanismos moleculares que intervienen en esta cardiogénesis anormal continúan siendo en gran parte desconocidos. Se acepta que las variaciones genéticas y epigenéticas son la causa predominante de las CC, si bien la identificación de las alteraciones precisas ha resultado difícil, principalmente porque la CC es un proceso complejo2.
Se ha identificado que los genes que participan en los controles transcripcionales, denominados factores de transcripción, desempeñan un papel importante en el desarrollo cardiaco3,4. Concretamente, se ha señalado que el factor de transcripción GATA4 es crucial para la especificación y el desarrollo normales del corazón5,6. Por lo que respecta a los demás factores de transcripción, se ha identificado una larga lista de mutaciones del gen GATA4 en los pacientes con CC, pero la contribución que cada una de estas mutaciones hace al riesgo de la enfermedad, sobre todo en lo relativo a las formas esporádicas, es muy pequeña y no está bien definida2,7,8. Recientemente, en estudios experimentales se ha puesto de manifiesto que los microácidos ribonucleicos (miARN) (moléculas de ARN pequeñas, de ∼20-22 nucleótidos, que no codifican proteínas) pueden modular la cardiogénesis alterando la expresión de esenciales proteínas reguladoras cardiacas7,9,10. En consonancia con estas observaciones, los datos de nuestro grupo han indicado que los frecuentes polimorfismos de nucleótido único (SNP) que se dan en la región 3’UTR del gen GATA4 alteran la unión de miARN-mARN y con ello causan una desregulación de la expresión del gen GATA411. El objetivo del presente trabajo es ampliar el análisis de la región 3’UTR del gen GATA4 mediante el análisis de SNP seleccionados y de haplotipos relacionados en esta región, con objeto de confirmar su importante papel en la modulación del riesgo de CC.
MÉTODOSPoblación en estudioFormaron la población en estudio 146 pacientes caucásicos (73 varones; edad, 6,68 ± 7,79 años) a los que se había diagnosticado una CC no sindrómica aislada y el grupo de control, 265 recién nacidos sanos (147 varones). Se obtuvo una muestra de sangre venosa de los participantes adultos, mientras que de los recién nacidos (tanto los que tenían una CC como los controles) se obtuvo una muestra de sangre del cordón umbilical. Este estudio se llevó a cabo tras obtener el consentimiento informado de todos los participantes o sus progenitores, y fue aprobado por el Comité de Ética de Investigación local.
Determinación del genotipoSe aisló ADN genómico a partir de las muestras de sangre utilizando métodos estándar, según las instrucciones del fabricante (QIAGEN BioRobot EZ1 System). Se amplificó la secuencia de la región 3’UTR mediante reacción en cadena de polimerasa (PCR) empleando cebadores específicos según un método descrito con anterioridad12. Los productos de la PCR se utilizaron para las reacciones de secuenciación de PCR mediante el CEQ DTCS Quick Start Kit. Después de la purificación, los productos de las reacciones de secuenciación se analizaron con una secuencia capilar CEQ 8800 (Beckman Coulter; Alemania) siguiendo el protocolo del fabricante. Las secuencias resultantes se analizaron empleando el software CEQ 8800 y se alinearon frente a una secuencia de referencia obtenida del banco génico Gen Bank BLAST (Basic Local Alignment Search Tool).
Selección del polimorfismo de nucleótido únicoLas 10 variantes genéticas frecuentes localizadas en la región 3’UTR del gen GATA4 que se observaron en nuestra población se analizaron para determinar presuntos lugares de unión de miARN empleando algoritmos de bioinformática, con objeto de calcular la diferencia de energía de hibridación libre (ΔFE, en kcal/mol) para cada alelo de tipo natural (wild-type) frente a la variante alélica, según un método descrito con anterioridad11,13. De manera resumida, se utilizó el MicroSNiPer14 para predecir la repercusión de cada SNP en las presuntas dianas de miARN. Los dúplex de miARN/diana muy estables se caracterizan por tener una energía libre mínima (kcal/mol) muy baja, que se ha calculado tanto para los alelos comunes como para las variantes alélicas mediante el programa RNACofold15, del paquete Vienna RNA (versión 1.8.5). La diferencia de energía libre entre los 2 alelos se calculó como «variación de FE» (ΔFE). Se calculó la suma de todos los ΔFE (|ΔFEtot|) para predecir la importancia biológica de los SNP que se unían a un mayor número de miARN.
Análisis estadísticoSe llevaron a cabo pruebas de asociación de locus único entre las frecuencias de alelos de los SNP y el carácter de caso-control mediante la prueba estándar de la t de Student para datos no apareados y el análisis de la χ2, utilizando para ello el paquete informático estadístico StatView, versión 5.0.1 (Abacus Concepts; Berkeley, California). Se aplicó un análisis de regresión logística para estimar la odds ratio (OR) y el intervalo de confianza del 95% (IC95%) correspondientes a la asociación entre la CC y la presencia del polimorfismo. En este análisis, se aplicó una corrección de Bonferroni para multiplicidad de pruebas (4 genotipos) en la evaluación de la significación estadística con un umbral del valor de p ajustado (p = 0,05 / 4 ≤ 0,0125).
Se realizaron pruebas de evaluación del equilibrio de Hardy-Weinberg para todos los loci en casos y controles por separado. Las medidas del desequilibrio de ligamiento, designadas como D’ y r2, y posterior determinación de las frecuencias de los haplotipos, se calcularon con los programas informáticos SNPAnalyzer 2.016 y SNP Stats17. Para el análisis de haplotipos, se empleó el algoritmo basado en la maximización de la expectativa18 y el algoritmo de ligadura de partición19, que permiten superar la existencia de datos de información de fase no disponibles mediante el examen de la fase de los polimorfismos de GATA4 y generar las estimaciones de máxima probabilidad de frecuencias de genotipos20.
Se consideró significativa una asociación si el valor de p bilateral era < 0,05.
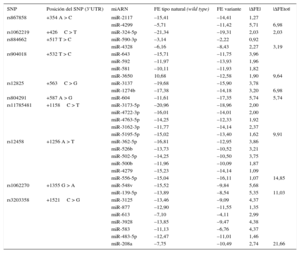
RESULTADOSLas características demográficas y clínicas de la población en estudio se muestran en la tabla 1. Cuatro SNP, +1158C > T (rs11785481), +1256 A > T (rs12458), +1355 G > A (rs1062270) y +1521C > G (rs3203358), situados en una región de 970 pb, mostraron los valores más altos de |ΔFEtot| (tabla 2). Se satisfizo la HWE respecto a cada polimorfismo analizado.
Características de la población en estudio
| Características | CC (n = 146) | Controles (n = 265) |
|---|---|---|
| Edad (años) | 6,68 ± 7,79 | 0 ± 0 |
| Varones/mujeres | 73/73 | 147/118 |
| Diagnóstico | ||
| Cardiopatía congénita cianótica | 56 | |
| Defecto del tabique | 52 | |
| Lesión obstructiva en el lado izquierdo | 6 | |
| Lesión mixta | 30 | |
| Ventrículo único | 2 | |
CC: cardiopatías congénitas.
Los valores expresan n o media ± desviación estándar.
ΔFE y ΔFEtot para los SNP de la región 3’UTR del gen GATA4
| SNP | Posición del SNP (3’UTR) | miARN | FE tipo natural (wild type) | FE variante | |ΔFE| | |ΔFEtot| |
|---|---|---|---|---|---|---|
| rs867858 | +354 A > C | miR-2117 | –15,41 | –14,41 | 1,27 | |
| miR-4299 | –5,71 | –11,42 | 5,71 | 6,98 | ||
| rs1062219 | +426C > T | miR-324-5p | –21,34 | –19,31 | 2,03 | 2,03 |
| rs884662 | +517 T > C | miR-590-3p | –3,14 | –2,22 | 0,92 | |
| miR-4328 | –6,16 | –8,43 | 2,27 | 3,19 | ||
| rs904018 | +532 T > C | miR-643 | –15,71 | –11,75 | 3,96 | |
| miR-592 | –11,97 | –13,93 | 1,96 | |||
| miR-581 | –10,11 | –11,93 | 1,82 | |||
| miR-3650 | 10,68 | –12,58 | 1,90 | 9,64 | ||
| rs12825 | +563C > G | miR-3137 | –19,68 | –15,90 | 3,78 | |
| miR-1274b | –17,38 | –14,18 | 3,20 | 6,98 | ||
| rs804291 | +587 A > G | miR-604 | –11,61 | –17,35 | 5,74 | 5,74 |
| rs11785481 | +1158C > T | miR-3173-5p | –20,96 | –18,96 | 2,00 | |
| miR-4722-3p | –16,01 | –14,01 | 2,00 | |||
| miR-4763-5p | –14,25 | –12,33 | 1,92 | |||
| miR-3162-3p | –11,77 | –14,14 | 2,37 | |||
| miR-5195-5p | –15,02 | –13,40 | 1,62 | 9,91 | ||
| rs12458 | +1256 A > T | miR-362-5p | –16,81 | –12,95 | 3,86 | |
| miR-526b | –13,73 | –10,52 | 3,21 | |||
| miR-502-5p | –14,25 | –10,50 | 3,75 | |||
| miR-500b | –11,96 | –10,09 | 1,87 | |||
| miR-4279 | –15,23 | –14,14 | 1,09 | |||
| miR-556-5p | –15,04 | –16,11 | 1,07 | 14,85 | ||
| rs1062270 | +1355 G > A | miR-548v | –15,52 | –9,84 | 5,68 | |
| miR-139-5p | –13,89 | –8,54 | 5,35 | 11,03 | ||
| rs3203358 | +1521C > G | miR-3125 | –13,46 | –9,09 | 4,37 | |
| miR-877 | –12,90 | –11,55 | 1,35 | |||
| miR-613 | –7,10 | –4,11 | 2,99 | |||
| miR-3928 | –13,85 | –9,47 | 4,38 | |||
| miR-583 | –11,13 | –6,76 | 4,37 | |||
| miR-483-5p | –12,47 | –11,01 | 1,46 | |||
| miR-208a | –7,75 | –10,49 | 2,74 | 21,66 |
3’UTR: región no traducida 3’; FE: energía libre; SNP: polimorfismo de nucleótido único; ΔFE: diferencia de energía de hibridación libre.
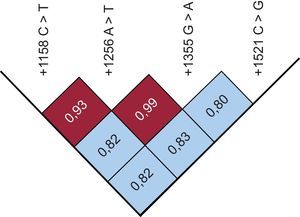
La distribución de los genotipos de las variantes +1158C > T y +1521C > G fue significativamente diferente en los casos y los controles. Concretamente, las frecuencias de +1158 CC, CT y TT fueron del 84, el 15 y el 1% de los pacientes en comparación con el 73, el 24 y el 3% de los controles (p < 0,04), mientras que las frecuencias de los genotipos +1521 CC, CG y GG fueron del 59, el 33 y el 8% de los pacientes en comparación con el 51, el 35 y el 14% de los controles (p < 0,05). Aunque el valor del desequilibrio de ligamiento emparejado (D’), corregido respecto a las frecuencias de los alelos (r2), mostró que los loci estaban en un desequilibrio intenso (figura), no se observaron diferencias significativas en la distribución de los genotipos y la frecuencia de los alelos entre los casos y los controles por lo que respecta a las variantes +1256 A > T y +1355 G > A.

El análisis de regresión logística reveló que el alelo T mutado del SNP +1158C > T y el alelo G del SNP +1521C > G se asociaban a una disminución del riesgo de CC, en modelos genéticos dominante y recesivo respectivamente (respectivamente, OR = 0,44; IC95%, 0,23-0,84; p = 0,01; y OR = 0,57; IC95%, 0,35-0,94; p = 0,03). No obstante, al aplicar una corrección por multiplicidad de pruebas, tan solo el alelo +1158 T mostró una diferencia significativa entre los pacientes y los controles.
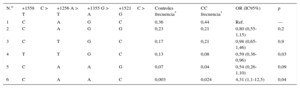
El análisis de los haplotipos puso de manifiesto 6 asociaciones de haplotipos en el grupo de casos y controles (tabla 3). El haplotipo T-T-G-C (el 8% de los casos de CC y el 13% del grupo de control) mostró un efecto protector en cuanto a la aparición de CC (OR = 0,59; IC95%, 0,36-0,96; p = 0,03) en comparación con el haplotipo más frecuente C-A-G-C. Tiene interés señalar que el haplotipo C-A-A-C, que era muy poco frecuente en los controles (0,3%) en comparación con los participantes que tenían una CC (2,4%), se asoció a un aumento de aproximadamente 4 veces en el riesgo de CC (OR = 4,31; IC95%, 1,1-12,5; p = 0,04).
Distribución de haplotipos de los 4 polimorfismos de GATA4 investigados en los casos de cardiopatía congénita y los controles
| N.o | +1558C > T | +1256 A > T | +1355 G > A | +1521C > G | Controles frecuencia* | CC frecuencia* | OR (IC95%) | p |
|---|---|---|---|---|---|---|---|---|
| 1 | C | A | G | C | 0,36 | 0,44 | Ref. | — |
| 2 | C | A | G | G | 0,23 | 0,21 | 0,80 (0,55-1,15) | 0,2 |
| 3 | C | T | G | C | 0,17 | 0,21 | 0,98 (0,65-1,46) | 0,9 |
| 4 | T | T | G | C | 0,13 | 0,08 | 0,59 (0,36-0,96) | 0,03 |
| 5 | C | A | A | G | 0,07 | 0,04 | 0,54 (0,26-1,10) | 0,09 |
| 6 | C | A | A | C | 0,003 | 0,024 | 4,31 (1,1-12,5) | 0,04 |
CC: cardiopatía congénita; IC95%: intervalo de confianza del 95%; OR: odds ratio.
El presente estudio confirma el importante papel que desempeña la región 3’UTR del gen GATA4 como factor de riesgo de CC. De hecho, las variantes genéticas frecuentes de esta región pueden interaccionar de manera conjunta, y afectar con ello la susceptibilidad a la CC y probablemente alterar el control postranscripcional del miARN. Además, hasta donde sabemos, este es el primer estudio que ha logrado identificar un locus de la región 3’UTR del GATA4 que tiene una utilidad potencial como biomarcador molecular para un diagnóstico precoz de las CC.
Se sabe que el factor de transcripción GATA4 es un regulador crucial de la expresión génica y la actividad celular en el corazón embrionario y posnatal6,21,22. El gen GATA4 contiene el código correspondiente a un componente de la familia de proteínas de unión GATA que se expresa en el endodermo del saco vitelino y el corazón embrionario que regula genes que intervienen posteriormente y son cruciales para la diferenciación y la función del miocardio. El GATA4 actúa de manera asociada a otros factores de transcripción, como NKX2-5 y TBX5, en un complejo transcripcional específico que confiere la expresión de genes específicos tisulares durante la cardiogénesis23. Las deleciones y mutaciones puntuales de GATA4, así como las duplicaciones del gen, se han asociado a CC24–26, si bien su frecuencia es muy baja, entre 0 y el 3%8,27. Hay un conjunto de evidencias que indican un papel importante de las modificaciones epigenéticas en los genes de factores de transcripción, incluida la regulación postranscripcional de los miARN. Hasta donde sabemos, no existen estudios sobre la expresión del perfil de miARN en el tejido cardiaco durante su desarrollo. Hay diferentes instrumentos bioinformáticos (como ESAdb o miRbase) que son capaces de predecir la expresión de miARN en diversos tejidos. El perfil de miARN tiene una naturaleza dinámica que se ve influida por múltiples factores, entre los que se encuentran la edad y las condiciones ambientales. De modo análogo, la regulación de la diana está bajo la influencia de mecanismos específicos espaciales y temporales. El tipo de célula, el estado de diferenciación de la célula y que la célula esté sometida a estrés o no son factores que parecen influir en que el miARN regule una diana o no28. Así pues, serán necesarios nuevos estudios ad hoc para identificar la expresión del perfil de miARN durante las primeas fases del desarrollo del corazón. Recientemente, hemos puesto de manifiesto que el miR-583 va dirigido específicamente al mARN de GATA4 y que, de manera más específica, los SNP frecuentes localizados en la región 3’UTR afectan a la regulación del gen GATA4 dependiente de miARN. De hecho, en células con transfección del alelo de tipo natural (wild) del +1521 C de la región 3’UTR de GATA4, el miR-583 redujo la actividad de luciferasa11. Por el contrario, no se detectó efecto alguno en las células con transfección del alelo mutante del +1521 G. Esto se debía a que un miARN de una longitud de 20–25 nucleótidos se une a un lugar diana de la región 3’UTR a través de la complementariedad de su región de semilla que incluye 2–8 nucleótidos29. En consecuencia, los SNP de la región 3’UTR correspondientes a la región de semilla pueden afectar a la fuerza de unión de un miARN específico, de tal manera que un alelo puede reducir, eliminar o crear la unión que modula la expresión génica11,13. Como resultado de ello, de manera similar a lo que ocurre con las «variantes funcionales» exónicas, las variantes situadas en regiones genómicas reguladoras pueden modificar también profundamente la expresión de los genes. En este estudio, se observa que otros 3 SNP (+1158C > T, +1355 G > A y +1256 A > T) que tienen los valores más altos de |ΔFEtot| predichos están situados muy cerca del SNP +1521C > G. Una región específica, situada en el extremo de la región 3’UTR del gen GATA4 que cubre 970 pb, podría ser la más sensible a la regulación por el miARN. Se confirmó que 2 de estos SNP (que muestran una heredabilidad conjunta en el 80% de las veces), +1158C > T y +1521C > G, mostraban una asociación independiente con la susceptibilidad a la CC. Aparentemente, los otros 2 SNP no presentaban asociación directa con la CC pese a estar en desequilibrio de ligamiento con los otros 2. El efecto de estos SNP en el fenotipo como marcadores aislados parece desdeñable, pero se manifiesta un efecto sinérgico en conjunto con los otros SNP. De hecho, el análisis de los haplotipos mostró claramente que el haplotipo T-T-G-C se asociaba a disminución del riesgo de CC. Por el contrario, el haplotipo C-A-A-C pudo aumentar este riesgo en 4 veces, lo cual confirma que estas 4 variantes de la región 3’UTR del GATA4 intervenían de manera sinérgica en la etiopatogenia de la CC de modo específico según el haplotipo en vez de como variante genética aislada.
Aunque nuestras observaciones respaldan claramente que hay una asociación bien definida entre la región 3’UTR de GATA4 y el riesgo de CC de un individuo, deben interpretarse teniendo en cuenta al menos 3 limitaciones importantes. En primer lugar, el estudio no tiene la potencia estadística suficiente para examinar la relación entre el haplotipo y el riesgo de CC. De hecho, incluso para el haplotipo que mostraba la asociación más intensa (0,13 frente a 0,08; p = 0,03), sería necesaria una población en estudio de 589 pacientes y 589 controles para disponer de una potencia estadística de 80. En segundo lugar, el estudio no dispuso de una validación externa en una población independiente. En tercer lugar, no realizamos análisis in vitro para evaluar el papel específico de los diferentes contextos de haplotipos y de los distintos miARN. A pesar de estas limitaciones, este estudio indica un papel importante de esta región genética y proporciona un punto de partida para un trabajo más amplio en este campo.
CONCLUSIONESNuestros datos muestran que ciertas variantes frecuentes de la región específica de 3’UTR de GATA4 tienen la capacidad de influir en la susceptibilidad a la CC, probablemente a través de una modificación de la regulación génica postranscripcional de los miARN. Serán necesarios nuevos estudios de mayor tamaño muestral para confirmar las repercusiones clínicas de la región 3’UTR de GATA4 como factor genético de riesgo de diagnóstico precoz de CC. Además, será útil llevar a cabo estudios in vitro sobre el efecto de ciertas variantes frecuentes de zonas de unión de miARN para definir mejor la regulación postranscripcional de la expresión génica de factores de transcripción por los miARN en la cardiogénesis.
CONFLICTO DE INTERESESNinguno.
- -
La cardiopatía congénita (CC) es el defecto neonatal más prevalente
- -
Se ha señalado que el factor de transcripción GATA4 es crucial para la especificación y el desarrollo normales del corazón
- -
Los miARN (moléculas de ARN de 20-22 nucleótidos que no codifican proteínas) pueden modular la cardiogénesis modificando la expresión de proteínas reguladoras cardiacas cruciales
- -
Los polimorfismos de nucleótido único (SNP) frecuentes de la región 3’UTR del gen GATA4 pueden alterar la unión de miARN-mARN, con lo que producen una disregulación de la expresión génica del GATA4
- -
Los alelos mutantes de los polimorfismos de GATA4 +1158 T y +1521 G mostraron asociación significativa con una reducción del riesgo de CC
- -
El haplotipo T-T-G-C (más infrecuente en los pacientes con CC que en los controles) se asoció a una disminución significativa del riesgo de CC, mientras que el haplotipo muy infrecuente C-A-A-C, que se daba de manera muy poco común en el grupo de control, se asoció a un aumento de 4 veces en el riesgo de enfermedad
- -
La región 3’UTR de GATA4 puede ser un locus genético de posible utilidad como biomarcador molecular para el diagnóstico precoz de CC
Los autores desean expresar su agradecimiento a cada una de las enfermeras y médicos de la Fondazione G. Monasterio CNR, Regione Toscana, por su continuo apoyo a este estudio. Damos las gracias también a nuestros pacientes y sus familias.