Single-nucleotide polymorphisms within a microRNA binding site can have different effects on gene expression, influencing the risk of disease. This study aimed to evaluate the association between single-nucleotide polymorphisms and haplotypes in the 3’UTR of the GATA4 gene and congenital heart disease risk.
MethodsBioinformatics algorithms were used to analyze single-nucleotide polymorphisms in putative microRNA-binding sites of GATA4 3’UTR and to calculate the difference in free energy of hybridization (ΔFE, kcal/mol) for each wild-type vs the variant allele.
ResultsThe study population comprised 146 Caucasian patients (73 males; 6.68 ± 7.79 years) and a 265 healthy newborn participants (147 males). The sum of all |ΔFE| was considered to predict the biological importance of single-nucleotide polymorphisms binding more microRNAs. Next, the 4 polymorphisms (+1158C > T, +1256 A > T, +1355 G > A, +1521C > G) with the highest predicted |ΔFEtot| (9.91, 14.85, 11.03, 21.66kcal/mol, respectively) were genotyped in a case-control study (146 patients and 250 controls). Applying a correction for multiple testing only the +1158 T allele was found to be associated with a reduced risk showing significant difference between patients and controls. Haplotype analysis showed that the T-T-G-C haplotype (more uncommon in congenital heart diseases than in controls) was associated with a significantly decreased risk (P = .03), while the rare C-A-A-C haplotype, which was very uncommon in controls (0.3%) compared with the disease (2.4%), was associated with a 4-fold increased risk of disease (P = .04).
ConclusionsCommon variants in 3’UTR of the GATA4 gene jointly interact, affecting the congenital heart disease susceptibility, probably by altering microRNA posttranscriptional regulation.
Keywords
Congenital heart disease (CHD) is the most prevalent of all birth defects (between 75 and 90 per 10 000 for live births in the last 20 years) and is the leading cause of death from congenital malformations in the neonatal period and during the first year of life.1 CHD comprises a heterogeneous group of cardiac defects that arise during fetal development. To date, the molecular mechanisms involved in such abnormal cardiogenesis remain largely unknown. Genetic and epigenetic variations are recognized as the predominant cause of CHD, although the identification of precise alterations has proven challenging, principally because CHD is a complex process.2
Genes involved in transcriptional controls, known as transcription factors, have been identified as major players in cardiac development.3,4 In particular, the transcription factor GATA4 is suggested to be crucial for normal heart specification and development.5,6 As for the other transcription factors, a long list of mutations in the GATA4 gene has been identified in CHD patients, but the contribution of each of these mutations to disease risk, especially for sporadic forms, is very low and not well-defined.2,7,8 Recently, experimental studies showed that miRNAs (nonprotein-coding small molecules of ribonucleic acid ∼20–22 nucleotides) may modulate cardiogenesis by altering the expression of critical cardiac regulatory proteins.7,9,10 Accordingly, data from our group indicated that common single nucleotide polymorphism (SNPs) in the 3’UTR of GATA4 gene altered the miRNA-mRNA binding, dysregulating GATA4 gene expression.11 The purpose of the present work was to expand the analysis of 3’UTR of the GATA4 gene by analyzing selected SNPs and related haplotypes in this region in order to confirm its major role in modulating CHD risk.
MethodsStudy PopulationThe study population comprised a group of 146 Caucasian patients (73 males; 6.68 ± 7.79 years), who were diagnosed with an isolated, nonsyndromic CHD and a control group of 265 healthy newborn participants (147 males). A sample of venous blood was collected from adult participants, whereas a cord blood sample was obtained from newborns (both CHDs and controls). This study was conducted with informed consent of all participants or their parents and was approved by the local Ethics Research Committee.
GenotypingGenomic DNA was isolated from blood using standard procedures according to the manufacturer's instructions (QIAGEN BioRobot EZ1 System). The 3’UTR sequence was amplified by polymerase chain reaction (PCR) using specific primers as previously described.12 The PCR products were used for PCR sequencing reactions by using the CEQ DTCS Quick Start Kit. After purification, the sequencing reaction products were analyzed with a CEQ 8800 capillary sequence (Beckman Coulter, Germany), according to the manufacturer's protocol. Resulting sequences were analyzed by using CEQ 8800 software packages and aligned against a reference sequence obtained from Gene Bank BLAST (Basic Local Alignment Search Tool).
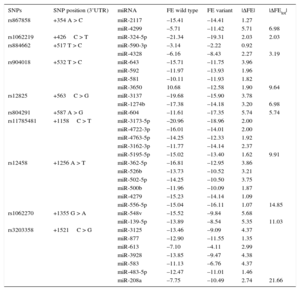
Single Nucleotide Polymorphism SelectionThe 10 common genetic variants located in the 3’UTR region of the GATA4 gene observed in our population were analyzed for putative miRNA-binding sites using bioinformatics algorithms in order to calculate the difference in free energy of hybridization (ΔFE, kcal/mol) for each wild-type vs variant allele, as previously described.11,13 Briefly, MicroSNiPer14 was used to predict the impact of each SNP on putative miRNA targets. Highly stable miRNA/target duplexes are represented as having a very low minimum free energy (kcal/mol) that has been calculated for both the common and the variant alleles by RNACofold program,15 from the Vienna RNA package (version 1.8.5). The difference in the free energies between the 2 alleles was computed as “variation of FE” (ΔFE). The sum of all |ΔFE| (|ΔFEtot|) was calculated to predict the biological importance of SNPs binding more miRNAs.
Statistical AnalysisSingle-locus tests of association between SNPs allele frequencies and case-control status were carried out via standard unpaired Student's t test and chi-square analysis, using StatView statistical package, version 5.0.1 (Abacus Concepts, Berkeley, California). Logistic regression analysis was used to estimate odds ratio (OR) and 95% confidence interval (95%CI) for the association between CHD and the presence of the polymorphism. In this analysis, a Bonferroni correction for (4 genotypes) multiple testing was performed to evaluate statistical significance at an adjusted P-value threshold (P = .05/4 ≤ .0125).
Tests for Hardy-Weinberg equilibrium were carried out for all loci among cases and controls separately. Measures of linkage disequilibrium, known as D’ and r2, and the subsequent evaluation of haplotype frequencies were computed by SNPAnalyzer 2.016 and SNP Stats softwares.17 The haplotype analysis was carried out using the expectation-maximization-based18 and the partition ligation algorithm19 that allow the missing phase information to be overcome by examining the phase of GATA4 polymorphisms and generating maximum likelihood estimates of haplotype frequencies.20
We considered an association to be significant if a 2-sided P-value was less than .05.
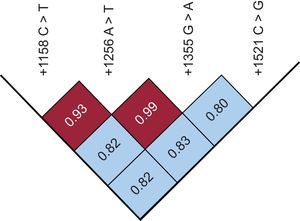
ResultsDemographic and clinical characteristics of the study population are shown in Table 1. Four SNPs, +1158C > T (rs11785481), +1256 A > T (rs12458), +1355 G > A (rs1062270), +1521C > G (rs3203358), located in a region of 970 pb, showed the highest |ΔFEtot| (Table 2). The HWE was satisfied for each polymorphism analyzed.
Characteristics of the Study Population
| Characteristics | CHD (n = 146) | Controls (n = 265) |
|---|---|---|
| Age, y | 6.68 ± 7.79 | 0 ± 0 |
| Male/female | 73/73 | 147/118 |
| Diagnosis, n | ||
| Cyanotic heart defect | 56 | |
| Septation defect | 52 | |
| Left-sided obstructive lesion | 6 | |
| Mixed lesion | 30 | |
| Single ventricle | 2 | |
CHD, congenital heart diseases.
Data are expressed as no. or mean ± standard deviation.
ΔFE and ΔFEtot for the 3’UTR SNPs of the GATA4 Gene
| SNPs | SNP position (3’UTR) | miRNA | FE wild type | FE variant | |ΔFE| | |ΔFEtot| |
|---|---|---|---|---|---|---|
| rs867858 | +354 A > C | miR-2117 | –15.41 | –14.41 | 1.27 | |
| miR-4299 | –5.71 | –11.42 | 5.71 | 6.98 | ||
| rs1062219 | +426C > T | miR-324-5p | –21.34 | –19.31 | 2.03 | 2.03 |
| rs884662 | +517 T > C | miR-590-3p | –3.14 | –2.22 | 0.92 | |
| miR-4328 | –6.16 | –8.43 | 2.27 | 3.19 | ||
| rs904018 | +532 T > C | miR-643 | –15.71 | –11.75 | 3.96 | |
| miR-592 | –11.97 | –13.93 | 1.96 | |||
| miR-581 | –10.11 | –11.93 | 1.82 | |||
| miR-3650 | 10.68 | –12.58 | 1.90 | 9.64 | ||
| rs12825 | +563C > G | miR-3137 | –19.68 | –15.90 | 3.78 | |
| miR-1274b | –17.38 | –14.18 | 3.20 | 6.98 | ||
| rs804291 | +587 A > G | miR-604 | –11.61 | –17.35 | 5.74 | 5.74 |
| rs11785481 | +1158C > T | miR-3173-5p | –20.96 | –18.96 | 2.00 | |
| miR-4722-3p | –16.01 | –14.01 | 2.00 | |||
| miR-4763-5p | –14.25 | –12.33 | 1.92 | |||
| miR-3162-3p | –11.77 | –14.14 | 2.37 | |||
| miR-5195-5p | –15.02 | –13.40 | 1.62 | 9.91 | ||
| rs12458 | +1256 A > T | miR-362-5p | –16.81 | –12.95 | 3.86 | |
| miR-526b | –13.73 | –10.52 | 3.21 | |||
| miR-502-5p | –14.25 | –10.50 | 3.75 | |||
| miR-500b | –11.96 | –10.09 | 1.87 | |||
| miR-4279 | –15.23 | –14.14 | 1.09 | |||
| miR-556-5p | –15.04 | –16.11 | 1.07 | 14.85 | ||
| rs1062270 | +1355 G > A | miR-548v | –15.52 | –9.84 | 5.68 | |
| miR-139-5p | –13.89 | –8.54 | 5.35 | 11.03 | ||
| rs3203358 | +1521C > G | miR-3125 | –13.46 | –9.09 | 4.37 | |
| miR-877 | –12.90 | –11.55 | 1.35 | |||
| miR-613 | –7.10 | –4.11 | 2.99 | |||
| miR-3928 | –13.85 | –9.47 | 4.38 | |||
| miR-583 | –11.13 | –6.76 | 4.37 | |||
| miR-483-5p | –12.47 | –11.01 | 1.46 | |||
| miR-208a | –7.75 | –10.49 | 2.74 | 21.66 |
3’UTR, 3’ untraslated region; FE, free energy; SNP, single nucleotide polimorphism; ΔFE, difference in free energy of hybridization.
The genotype distribution of +1158C > T and +1521C > G variants was significantly different between cases and controls. Specifically, the frequencies of +1158 CC, CT, and TT were 84%, 15%, and 1% in patients compared with 73%, 24%, and 3% in controls (P < .04), while the frequencies of +1521 CC, CG, and GG genotypes were 59%, 33%, and 8% in patients compared with 51%, 35%, and 14% in controls (P < .05). Although the pairwise linkage disequilibrium value (D’), corrected for allele frequencies (r2), showed that the loci were in strong disequilibrium (Figure), no significant differences in genotype distribution and allele frequency between cases and controls were observed for the +1256 A > T and +1355 G > A variants.

Logistic regression analysis revealed that the mutated T allele of +1158C > T SNP as well as the G allele of +1521C > G SNP were associated with a decreased risk for CHD, under a dominant and recessive genetic model, respectively (OR = 0.44; 95%CI, 0.23-0.84; P = .01; and OR = 0.57; 95%CI 0.35–0.94; P = .03; respectively). Nevertheless, when we applied a correction for multiple testing only, the +1158 T allele showed significant difference between patients and controls.
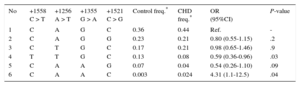
Haplotype analysis showed 6 haplotype associations in the case and control group (Table 3). The T-T-G-C haplotype (8% in CHD cases and 13% in the control group) had a protective role in the development of CHD (OR = 0.59; 95%CI, 0.36-0.96; P = .03) compared with the most common C-A-G-C haplotype. Interestingly, the C-A-A-C haplotype, which was very uncommon in controls (0.3%) compared with participants with CHD (2.4%), was associated with a roughly 4-fold increased risk of CHD (OR = 4.31; 95%CI, 1.1-12.5; P = .04).
Haplotype Distribution of the 4 Investigated GATA4 Polymorphisms in Congenital Heart Disease Cases and Controls
| No | +1558 C > T | +1256 A > T | +1355 G > A | +1521 C > G | Control freq.* | CHD freq.* | OR (95%CI) | P-value |
|---|---|---|---|---|---|---|---|---|
| 1 | C | A | G | C | 0.36 | 0.44 | Ref. | - |
| 2 | C | A | G | G | 0.23 | 0.21 | 0.80 (0.55-1.15) | .2 |
| 3 | C | T | G | C | 0.17 | 0.21 | 0.98 (0.65-1.46) | .9 |
| 4 | T | T | G | C | 0.13 | 0.08 | 0.59 (0.36-0.96) | .03 |
| 5 | C | A | A | G | 0.07 | 0.04 | 0.54 (0.26-1.10) | .09 |
| 6 | C | A | A | C | 0.003 | 0.024 | 4.31 (1.1-12.5) | .04 |
95%CI, 95% confidence interval; CHD, congenital heart disease; OR, odds ratio.
The present study confirms the major role of 3’UTR of the GATA4 gene as a risk factor for CHD. Indeed, common genetic variants in this region can jointly interact, affecting susceptibility to CHD and likely altering miRNA post-transcriptional control. Additionally, to our knowledge, this is the first study able to identify a GATA4 3’UTR locus potentially useful as molecular biomarkers for an early diagnosis of CHD.
The transcription factor GATA4 is known as a critical regulator of gene expression and cellular activity in the embryonic and postnatal heart.6,21,22 The GATA4 gene encodes a member of the GATA-binding protein family expressed in yolk sac endoderm and embryonic heart that regulates downstream genes critical for myocardial differentiation and function. GATA4 acts in association with other transcription factors, such as NKX2-5 and TBX5, in a specific transcriptional complex that confers tissue-specific gene expression during cardiogenesis.23 Deletions and point mutations of GATA4, as well as gene duplications, have been associated with CHD,24–26 although their frequency is very low, ranging from 0% to 3%.8,27 A series of evidence suggests a major role of epigenetic modifications in transcription factor genes including miRNA posttranscriptional regulation. To the best of our knowledge, there are no studies on the expression of miRNA profile in heart tissue during its development. Different bio-informatics tools (ie, ESAdb or miRbase) are able to predict miRNA expression in several tissues. The miRNA profile has a dynamic nature influenced by many factors, including age and environmental conditions. Similarly, target regulation is under the influence of temporal and spatial-specific mechanisms. The cell type, the differentiation state of the cell, and whether a cell is under stress all appear to influence whether a miRNA regulates a target.28 Thus, further ad-hoc studies are warranted to identify miRNA profile expression during the first stages of heart development. Recently, we showed that miR-583 specifically targeted GATA4 mRNA and that, more specifically, common SNPs located in the 3’UTR region affected miRNA-dependent GATA4 gene regulation. Indeed, in cells transfected with +1521C wild type allele of GATA4 3’UTR, miR-583 decreased luciferase activity.11 Conversely, no effect was detected in cells transfected with the +1521 G mutant allele. This was because a miRNA 20-25 nucleotides long binds a 3’UTR target site through the complementarity of its seed region that includes 2-8 nucleotides.29 Therefore, SNPs in the 3’UTR region corresponding to the seed region may affect the bond strength of a specific miRNA so that 1 allele may reduce, eliminate or create the binding that modulates gene expression.11,13 As result, similarly to exonic “functional variants”, variants localized in regulatory genomic regions can also deeply alter gene expressions. In this study, we found that 3 other SNPs (+1158C > T, +1355 G > A, +1256 A > T) with the highest predicted |ΔFEtot|s, are located very close to the +1521C > G SNP. A specific region, in the 3’UTR extremity of the GATA4 gene covering 970bp, might be the most sensitive region to miRNA regulation. Two of these SNPs (which were co-inheritable 80% of the time), +1158C > T and +1521C > G, were confirmed to be independently associated with CHD susceptibility. Apparently, the other 2 SNPs did not associate directly with CHD, despite being in linkage disequilibrium with the other 2. The effect of these SNPs on phenotype as a single biomarker seems to be negligible but they exhibit their effect synergistically in conjunction with the other SNPs. Indeed, the haplotype analysis clearly showed that the T-T-G-C haplotype was associated with a decreased risk of CHD. Conversely, the C-A-A-C haplotype was able to increase this risk by 4-fold, confirming that these 4 GATA4 3’UTR variants were synergistically involved in the etiopathogenesis of CHD in a haplotype-specific fashion rather than as single genetic variant.
Although our findings strongly support a well-defined association between GATA4 3’UTR and individual CHD risk, they should be interpreted bearing in mind at least 3 important limitations. First, the study is underpowered in examining the relationship between haplotype and CHD risk. Indeed, even for the haplotype with the strongest association (0.13 vs 0.08; P = .03), a study population of 589 patients and 589 controls would be required to have a power of 80. Second, the study lacked external validation in an independent population. Third, we did not perform any in vitro analyses to assess the specific roles of different haplotype settings and miRNAs. Despite these limitations, this study suggests a major role of this genetic region and offers a starting point for more extensive work in this field.
ConclusionsOur data showed that common variants in a specific region of GATA4 3’UTR are able to influence susceptibility to CHD, likely by altering the miRNA posttranscriptional gene regulation. Future studies with a large sample size are necessary to confirm the clinical impact of GATA4 3’UTR as a genetic risk factor for an early diagnosis of CHD. Furthermore, in vitro studies focused on the effect of common variants in miRNA binding sites will be useful to better define the posttranscriptional regulation of transcription factor gene expression by miRNAs in cardiogenesis.
Conflicts of InterestNone declared.
- -
Congenital heart disease (CHD) is the most prevalent of all birth defects.
- -
The transcription factor GATA4 is suggested to be crucial for normal heart specification and development.
- -
miRNAs (non-protein coding 20–22 nucleotides molecules of RNA) may modulate cardiogenesis by altering the expression of critical cardiac regulatory proteins.
- -
Common single nucleotide polymorphism (SNPs) in the 3’UTR of the GATA4 gene may alter miRNA-mRNA binding, dysregulating GATA4 gene expression.
- -
The mutant alleles of GATA4 +1158 T and +1521 G polymorphisms were significantly associated with a reduced risk of CHD.
- -
The T-T-G-C haplotype (more uncommon in congenital heart diseases than in control) was associated with a significantly decreased risk of CHD, while the rare C-A-A-C haplotype, which was very uncommon in the control group was associated with a 4-fold increased risk of disease.
- -
GATA4 3’UTR may be a genetic locus potentially useful as a molecular biomarker for an early diagnosis of CHD.
The authors wish to thank each nurse and physician at the Fondazione G. Monasterio CNR, Regione Toscana, for their continuous support in this study. We are also grateful to our patients and their families.