La displasia arritmogénica de ventrículo derecho (DAVD) es una enfermedad de etiología genética, con una frecuencia de mutaciones de un 50-60% en los genes desmosomales. En general, la herencia de la enfermedad es autosómica dominante, excepto en los casos de enfermedad de Naxos y Carvajal, con características fenotípicas concretas (queratodermia palmoplantar, pelo rizado, etc.) y con herencia autosómica recesiva. Desde que en 2006 se asociase por primera vez el gen de la desmogleína 2 (DSG2) con el desarrollo de DAVD1, han sido numerosas las mutaciones descritas, casi todas ellas heterocigotas con comportamiento autosómico dominante. En ocasiones este gen se hereda como heterocigosis compuesta o digénica2. Recientemente se ha señalado que las mutaciones en este gen se pueden comportar de forma autosómica recesiva en ausencia de síndrome cardiocutáneo, si bien los datos disponibles son muy escasos3,4. Nuestro objetivo es describir el espectro clínico y el modo de transmisión autosómico recesivo de una doble variante en DSG2 en ausencia de síndrome cardiocutáneo.
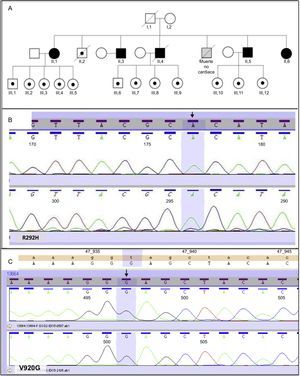
Se incluyó a 18 sujetos de una misma familia. El árbol familiar se muestra en la figura A; 5 pacientes eran homocigotos para las mutaciones R292H y V920G en DSG2, y todos pertenecían a la segunda generación. Los otros 13 eran heterocigotos para ambas mutaciones, y 12 de ellos pertenecían a la tercera generación familiar, mientras que uno (sujeto II.2) se encontraba en la segunda generación. La mutación R292H (figura B) en DSG2 condiciona un cambio aminoacídico de arginina por histidina en el tercer dominio extracelular de la región amino N-terminal. La segunda variante condiciona un cambio aminoacídico de valina por glicina (figura C) en el extremo C-terminal citoplasmático3.
. C: variante homocigota V920G en DSG2 (flecha). Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
A: árbol familiar que muestra a los sujetos homocigotos en negro y a los portadores heterocigotos, blanco con un punto negro. Los cuadros simbolizan a los varones y los círculos, a las mujeres. B: variante homocigota R292H en DSG2 (flecha). C: variante homocigota V920G en DSG2 (flecha). Esta figura se muestra a todo color solo en la versión electrónica del artículo.
De todos los sujetos, se recogió información clínica relativa a eventos como síncope, arritmias ventriculares, descargas del desfibrilador automático, hospitalización por insuficiencia cardiaca y clase funcional. Se estudió a todos con electrocardiograma de 12 derivaciones, ecocardiograma y, cuando fue posible, resonancia magnética cardiaca y Holter de 24h.
El estudio estadístico se realizó comparando la presencia de ambas variantes en homocigosis o heterocigosis. Para investigar diferencias en eventos clínicos, empleamos el test exacto de Fisher con el programa estadístico SPSS 19.0 (Chicago, Estados Unidos).
La media de edad de los sujetos homocigotos fue superior que la de los heterocigotos, debido a que todos pertenecían a la segunda generación, mientras que solo 1 de los 13 heterocigotos era de esta generación (sujeto II.2 en la figura A). No hubo diferencias en cuanto a sexo.
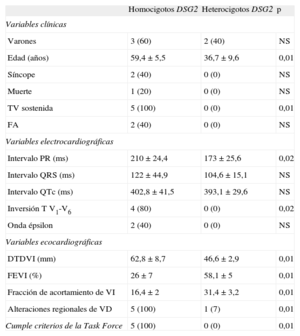
Todos los sujetos homocigotos mostraron un cuadro clínico compatible con displasia arritmogénica con afección biventricular, arritmias ventriculares graves y función ventricular izquierda muy afectada. Esto fue estadísticamente diferente respecto a los sujetos heterocigotos, de los que ninguno cumplía criterios diagnósticos de DAVD. Ninguno de los 5 casos homocigotos presentó rasgos fenotípicos característicos de síndrome de Naxos o Carvajal, y en todos los casos precisaron el implante de desfibrilador automático. La comparación estadística de datos clínicos, electrocardiográficos y ecocardiográficos entre ambos grupos se muestra en la tabla.
Comparación estadística de datos clínicos entre homocigotos y heterocigotos
| Homocigotos DSG2 | Heterocigotos DSG2 | p | |
| Variables clínicas | |||
| Varones | 3 (60) | 2 (40) | NS |
| Edad (años) | 59,4±5,5 | 36,7±9,6 | 0,01 |
| Síncope | 2 (40) | 0 (0) | NS |
| Muerte | 1 (20) | 0 (0) | NS |
| TV sostenida | 5 (100) | 0 (0) | 0,01 |
| FA | 2 (40) | 0 (0) | NS |
| Variables electrocardiográficas | |||
| Intervalo PR (ms) | 210±24,4 | 173±25,6 | 0,02 |
| Intervalo QRS (ms) | 122±44,9 | 104,6±15,1 | NS |
| Intervalo QTc (ms) | 402,8±41,5 | 393,1±29,6 | NS |
| Inversión T V1-V6 | 4 (80) | 0 (0) | 0,02 |
| Onda épsilon | 2 (40) | 0 (0) | NS |
| Variables ecocardiográficas | |||
| DTDVI (mm) | 62,8±8,7 | 46,6±2,9 | 0,01 |
| FEVI (%) | 26±7 | 58,1±5 | 0,01 |
| Fracción de acortamiento de VI | 16,4±2 | 31,4±3,2 | 0,01 |
| Alteraciones regionales de VD | 5 (100) | 1 (7) | 0,01 |
| Cumple criterios de la Task Force | 5 (100) | 0 (0) | 0,01 |
DSG2: gen de desmogleína 2; DTDVI: diámetro telediastólico del ventrículo izquierdo; FA: fibrilación auricular; FEVI: fracción de eyección del ventrículo izquierdo; Task Force: Grupo de Trabajo en Enfermedades del Pericardio y el Miocardio de la Sociedad Europea de Cardiología y el Scientific Council on Cardiomyopathies de la International Society and Federation of Cardiology; TV: taquicardia ventricular; VD: ventrículo derecho; VI: ventrículo izquierdo.
Los datos expresan n (%) o media±deviación estándar.
Los 13 sujetos heterocigotos para ambas mutaciones mostraron electrocardiograma compatible con la normalidad y ausencias de eventos clínicos. Un caso heterocigoto de la segunda generación (sujeto II.2 en la figura A) presentaba cardiopatía isquémica, pero ningún dato que indicara displasia arritmogénica.
Nuestro trabajo es el primero que describe una doble variante homocigota con comportamiento autosómico recesivo en un gen desmosomal, en este caso DSG2, como causa de DAVD con expresividad clínica grave. Es interesante la ausencia de hallazgos típicos de los síndromes de Carvajal y Naxos, con lo que se extiende la evidencia de que la DAVD puede presentarse en ocasiones como una enfermedad autosómica recesiva con genotipo complejo, sin síndromes cardiocutáneos asociados. Estos resultados señalan la necesidad de ser prudentes en la interpretación de variantes heterocigotas sin fenotipo en la práctica real5.
De las 2 variantes homocigotas que describimos en nuestro trabajo, la mostrada en la figura B es una mutación no descrita, muy infrecuente en controles y con altas probabilidades de alterar la arquitectura proteica del desmosoma. La otra variante, V920G, que afecta a un residuo conservado entre especies y está ausente en más de 200 controles sanos, se ha descrito previamente como posible causa de miocardiopatía dilatada y arritmogénica, aunque asociada a otras variantes heterocigotas en el propio DSG2 u otro gen desmosomal como la desmoplaquina, sin quedar clara su patogenicidad por sí sola.
En conclusión, nuestro estudio describe un modo de transmisión autosómico recesivo, con 2 variantes homocigotas en DSG2, una de ellas claramente patogénica, como causa de DAVD con afección biventricular grave, en ausencia de signos clínicos de síndrome cardiocutáneo. No se puede afirmar con rotundidad que sea la interacción de ambas mutaciones la causante de este fenotipo recesivo tan marcado, que puede deberse en gran parte a un efecto patogénico de R292H y modulado por V920G.