Se presenta el caso de una familia con 8 miembros de 3 generaciones afectados por el síndrome de Marfan (SMF), en los que un completo estudio familiar y genético confirma la patogenicidad de una variante «sinónima» identificada en el gen de la fibrilina 1 (FBN1).
El caso índice es un varón caucásico de 50 años intervenido de urgencia por una disección de aorta tipo A. Su padre, de 79 años, había sido intervenido a los 56 años por dilatación de la aorta ascendente para sustitución de aorta y reemplazo valvular (técnica de Bentall) y se lo intervino de nuevo a los 75 años por dilatación de la aorta descendente. Al caso índice se le había detectado, años antes de su disección, una ectopia lentis y, dado el antecedente de su padre, se sospechó un posible SMF. En 2008 se le realizó un estudio genético del gen FBN1 en otro centro, mediante Sanger y multiplex ligation-dependent probe amplification (MLPA), pero se informó como negativo. Sus 2 hijos se estaban siguiendo en un tercer centro por sospecha de SMF.
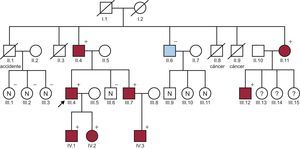
En nuestro hospital se centralizó el estudio familiar. Al elaborar el árbol genealógico (figura), se identificó a otros miembros con dilatación de la aorta o con una expresión variable de signos de SMF1 (tabla). El hermano menor, también con ectopia lentis, se intervino recientemente por un aneurisma de aorta ascendente, y se le implantó un tubo valvulado Bentall. Este paciente rechazó la cirugía de preservación del anillo aórtico ofrecida (técnica de David), debido a la buena evolución que su padre y su hermano habían tenido con la técnica de Bentall.

Árbol familiar. Cuadrado: varón; círculo: mujer; flecha: índice; rojo: afectado SMF; azul: aorta bicúspide; línea oblicua: fallecido; N: evaluado y sano; ?: desconocido; +: portador p.Ile2118=; –: no portador. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
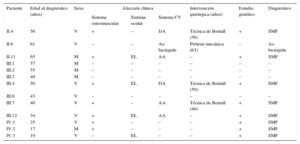
Información clínica de los miembros de la familia estudiada
| Paciente | Edad al diagnóstico (años) | Sexo | Afección clínica | Intervención quirúrgica (años) | Estudio genético | Diagnóstico | ||
|---|---|---|---|---|---|---|---|---|
| Sistema osteomuscular | Sistema ocular | Sistema CV | ||||||
| II.4 | 56 | V | + | – | DA | Técnica de Bentall (56) | + | SMF |
| II.6 | 61 | V | – | – | Ao bicúspide | Prótesis mecánica (63) | – | Ao bicúspide |
| II.11 | 65 | M | + | EL | AA | – | + | SMF |
| III.1 | 57 | M | – | – | – | – | – | – |
| III.2 | 55 | M | – | – | – | – | – | – |
| III.3 | 49 | M | – | – | – | – | – | – |
| III.4 | 50 | V | + | EL | DA | Técnica de Bentall (50) | + | SMF |
| III.6 | 43 | V | – | – | – | – | – | – |
| III.7 | 40 | V | + | – | AA | Técnica de Bentall (40) | + | SMF |
| III.12 | 34 | V | + | EL | AA | – | + | SMF |
| IV.1 | 25 | V | + | – | – | – | + | SMF |
| IV.2 | 17 | M | + | – | – | – | + | SMF |
| IV.3 | 19 | V | – | EL | – | – | + | SMF |
–: negativo; +: positivo; AA: aneurisma de aorta; Ao: aorta; CV: cardiovascular; DA: disección de aorta; EL: ectopia lentis; M: mujer; SMF: síndrome de Marfan; V: varón.
Sistema osteomuscular + en presencia de alguna de las siguientes alteraciones: aracnodactilia, escoliosis, deformidad torácica, estrías en piel, protrusión acetabular, pie plano o deformidad del pie.
Dada la evidente enfermedad familiar, llamaba la atención la negatividad del estudio genético realizado previamente. En el SMF se han descrito porcentajes de positividad del estudio genético cercanos al 90%2. Se decidió realizar un nuevo estudio, esta vez mediante next generation sequencing con un panel que incluía 30 genes implicados en enfermedades aórticas. El estudio únicamente identificó una variante «sinónima» (el cambio de nucleótido no produce cambio de aminoácido) en el gen FBN1 (c.6354C>T, p.Ile2118=). El hecho de que no se produzca un cambio de aminoácido podría indicar que esta variante es no patogénica o de dudosa patogenicidad. Está localizada en una región codificante del gen (en la posición 40 del exón 52, isoforma NM_000138.4), posición que no parece que afecte a ninguno de los sitios relevantes para el proceso de corte y empalme del ARN (proceso de splicing). Sin embargo, un estudio de expresión de ARN, llevado a cabo por Liu et al3. en 1977, mostró que dicho cambio genera un skipping del exón 52 (ausencia de este exón en el transcripto de ARN). Esto puede generar una proteína truncada por la creación de una nueva pauta de lectura que podría dar lugar a un codón de stop prematuro, o a que no se exprese la proteína FBN1 porque la maquinaria celular degrada el transcripto aberrante. El hecho de que el splicing esté afectado y que tenga efectos funcionales demostrados sería suficiente para demostrar la patogenicidad de la mutación4. Además, esta variante no aparece en bases de datos que incluyen información de población utilizada como control. Pero para confirmar definitivamente la patogenicidad, falta demostrar su cosegregación en alguna de las familias. A pesar de que se han publicado 8 casos de distintas familias con sospecha o diagnóstico clínico de SMF, en los artículos no se aportan datos del estudio familiar, únicamente de los casos índice5,6.
En la familia que se presenta, se ha demostrado por primera vez la cosegregación de esta mutación con la enfermedad. Todos ellos cumplían los nuevos criterios de Ghent4 (tabla). Solo hubo 1 caso (II.6) en el cual no se identificó la variante, un tío paterno del índice. Se confirmó que tenía una enfermedad no relacionada (aorta bicúspide) y se lo había intervenido por una estenosis aórtica. No se pudo estudiar a 3 hijas de un afectado (III.13,14,15) por problemas sociales.
En conclusión, con un completo estudio clínico y genético familiar, por primera vez, se ha podido establecer la cosegregación de la variante p.Ile2118= en el gen FBN1 con el SMF y confirmar con ello su patogenicidad. No sabemos si el estudio genético realizado años antes no identificó esta variante (falso negativo del Sanger) o se consideró no patogénica.
Es importante disponer de centros de referencia en SMF o unidades de cardiopatías familiares que permitan realizar estudios familiares completos2. El estudio familiar junto con una correcta interpretación de las variantes identificadas en los estudios genéticos permitirá establecer un pronóstico y una asistencia clínica más precisos.
FINANCIACIÓNEste trabajo ha sido parcialmente financiado por la Red de Investigación Cardiovascular (RIC; RD12/0042/0069).
CONFLICTO DE INTERESESJ.P. Trujillo-Quintero pertenece al departamento clínico de Health in Code.