
Palabras clave
Introducción
La miocardiopatía hipertrófica (MCH) es la causa más frecuente de muerte súbita cardiaca (MSC) en adultos jóvenes, y una causa importante de morbimortalidad a edad avanzada. El cuadro clínico asociado es muy variable y abarca desde los síntomas incapacitantes hasta la ausencia de síntomas. Muchos pacientes permanecen asintomáticos durante largos periodos, aunque el porcentaje de pacientes con síntomas severos aumenta con la edad1.
Se ha estimado2 que el 0,2% de las personas tendrían un grosor de la pared ≥ 15 mm. En la base fisiopatológica de la MCH están las mutaciones en los genes que codifican las proteínas del sarcómero. Se diagnostica a un 30-40% de los pacientes como casos esporádicos, pero la penetrancia incompleta de algunas mutaciones podría subestimar el porcentaje de los casos realmente familiares3. La heterogeneidad clínica se debe en primer lugar a la existencia de al menos 12 genes que pueden estar mutados. Las primeras mutaciones se hallaron en el gen MYH7, que codifica la cadena pesada de la betamiosina cardiaca4-7. Más tarde se identificaron mutaciones en otros genes, como TNNT2 (troponina T) y MYBPC3 (proteína C de unión a la miosina cardiaca)8-14.
La mayor parte de las mutaciones se han hallado en una sola familia, lo que dificulta la obtención de datos concluyentes sobre el fenotipo asociado a cada mutación. Sólo en las encontradas en un número grande de pacientes se han obtenido datos relevantes de la correlación genotipo-fenotipo. Los primeros estudios indicaban que las mutaciones en MYH7 darían formas severas de hipertrofia y en TNNT2, una hipertrofia menos severa pero elevado riesgo de MSC. Los portadores de mutaciones en MYBPC3 presentarían formas menos severas y menos riesgo de MSC3,8,12-25. Sin embargo, se han descrito mutaciones de mal pronóstico en genes inicialmente relacionados con formas menos agresivas, algo que ilustraría la dificultad de predecir el fenotipo a partir del genotipo. El objetivo de nuestro estudio es identificar la prevalencia y las características fenotípicas de las mutaciones en cinco genes sarcoméricos (MYH7, MYBPC3, TNNT2, TNNI3y TPM1) en pacientes de Asturias y Cantabria.
Métodos
Pacientes
Estudiamos a 120 pacientes no emparentados y diagnosticados entre 2002 y 2007 por especialistas de los servicios de cardiología del Hospital Universitario Central de Asturias y del Hospital Universitario Marqués de Valdecilla de Santander. El diagnóstico se realizó siguiendo los criterios de la ACC/ ESC, tomando como criterio de inclusión un grosor ecocardiográfico de la pared ventricular izquierda > 15 mm en ausencia de otras causas que justificasen la hipertrofia26. Las características clínicas de los pacientes se resumen en la tabla 1.

Se consideró casos familiares a los pacientes con algún pariente que también había sido diagnosticado de MCH y esporádicos a aquellos de los que no había constancia de otros familiares afectados. En los pacientes en los que se halló alguna mutación se determinó su presencia en todos los familiares que accedieron a participar en el estudio, independientemente de si tenían o no síntomas de la enfermedad, y a los que eran portadores de la mutación se les realizó un estudio ecocardiográfico.
Todos los participantes firmaron un consentimiento informado para ser incluidos en el estudio, que fue aprobado por el Comité Ético de Investigación Clínica del Hospital Universitario Central de Asturias.
Análisis genético
Mediante la reacción en cadena de la polimerasa, se amplificaron los exones y las bases intrónicas flanqueantes de los genes MYH7 (38 exones), MYBPC3(34 exones), TNNT2 (15 exones), TNNI3 (9 exones) y TPM1 (9 exones). Los cebadores empleados para la reacción en cadena de la polimerasa se diseñaron a partir de las secuencias de referencia depositadas en la base de datos GenBank. Cada fragmento de la reacción en cadena de la polimerasa se purificó y se secuenció mediante química de BigDye en un equipo ABI310 (Applied Biosystems; Foster City, California, Estados Unidos) (fig. 1). Las mutaciones y los polimorfismos hallados en los cinco genes sarcoméricos se nombraron siguiendo la base de datos Cardiogenomics (www.cardiogenomics.org). Los cebadores y las condiciones de amplificación serán facilitados por los autores en la dirección de correspondencia.

Fig. 1. A: patrones electroforéticos en geles SSCA para el fragmento del exón 13 del gen MYBPC3. Las calles 1, 2 y 6 corresponden a secuencias normales (sin mutaciones). En la calle 3 el patrón corresponde al cambio V342D (mutación); en la 4, al cambio R326Q (polimorfismo) y en la 5, a la deleción A328fs (mutación). B: secuencias de los fragmentos con las tres variantes nucleotídicas del exón 13 de MYBPC3 (*secuencia de la hebra inversa).
Las mutaciones en los genes TNNT2 (15 exones) y TNNI3 (9 exones) en 115 de los 120 casos y en exones seleccionados de MYH7 y MYBPC3 en algunos pacientes ya han sido publicados27-29.
Estudio en controles
Para considerar un cambio nucleotídico en un gen como una mutación asociada al desarrollo de una enfermedad, deben cumplirse varias condiciones. Primero, el cambio debe alterar la secuencia de aminoácidos de la proteína. Segundo, la mutación debe hallarse en los afectados de una misma familia, por lo que podríamos excluir el efecto patogénico de un cambio si algún afectado no lo hubiese heredado. Por otro lado, los portadores de la mutación tendrían mayor probabilidad de desarrollar la enfermedad, por lo que no deberían hallarse entre personas sin síntomas de la enfermedad.
Todas las variantes halladas en los pacientes y que no figuraban en las bases de datos como polimorfismos se analizaron en 200 personas sanas que habían firmado un consentimiento informado para participar en el estudio. Todos eran mayores de 18 años y no tenían síntomas de enfermedad cardiovascular, aunque no fueron estudiados ecocardiográficamente, por lo que no podemos descartar una hipertrofia asintomática. Cada fragmento en el que se halló un cambio nucleotídico que podría ser una mutación se amplificó en el paciente que lo presentaba y en los 200 controles, y se analizó su patrón de migración electroforética en geles de poliacrilamida no desnaturalizantes mediante la técnica SSCA y siguiendo un protocolo descrito previamente (fig. 1)27,28.
Grado de conservación de los aminoácidos entre especies
Las mutaciones afectan a aminoácidos importantes para la estructura y la función de la proteína, lo que limitaría su divergencia evolutiva. Todos los cambios nucleotídicos que modificaban la secuencia de la proteína y no fueron hallados en los controles se consideraron posibles mutaciones patogénicas. Como criterio adicional de implicación en la enfermedad, determinamos su grado de conservación entre el hombre, el chimpancé y el ratón, comparando las secuencias de las tres especies depositadas en la base de datos ENSEMBL (www.ensembl.org).
Análisis estadístico
Se empleó el programa estadístico SPSSTM versión 11.0 para los análisis estadísticos. Mediante ANOVA y la prueba de la U de Mann-Whitney, se compararon las variables continuas, y se empleó la prueba de la χ2 para las variables discretas. En todos los análisis se consideraron estadísticamente significativas las diferencias con una p < 0,05.
Resultados
En 109 (91%) pacientes, la sospecha de MCH se derivó de sus manifestaciones clínicas (disnea de esfuerzo, palpitaciones, angina o síncope) y en 11, por la existencia de alteraciones electrocardiográficas en un examen médico habitual. De los 120 pacientes, 35 (29%) tenían al menos un familiar que también padecía MCH. Se hallaron 31 mutaciones en 32 pacientes: 10 con mutación en MYH7, 20 en MYBPC3, 2 en TNNT2 y 1 en TPM1 (tablas 2 y 3). Dos mutaciones en MYBPC3 (G263X y E542Q) fueron halladas en más de 1 paciente, 1 tenía dos mutaciones (R278C-TNNT2 y R733H-MYBPC3) y otro era homocigoto para la mutación A627V en MYBPC3.


Mutaciones en MYH7
De los 120 pacientes, 10 (8%) tenían mutaciones en el gen de la cadena pesada de la betamiosina (tabla 2). Nueve mutaciones se localizaban en los primeros 22 exones del gen y solamente el cambio K1459N en la cola de la proteína. Cuatro de las mutaciones habían sido descritas. La media de edad al diagnóstico en estos 10 pacientes fue 35 años (tabla 1). Siete (70%) tenían antecedentes familiares de la enfermedad. Los pacientes con R453C, A583V y R663H tenían familiares que habían sufrido MSC precoz (antes de los 50 años). La mutación V822M se halló en una mujer diagnosticada a la edad de 3 años. Esta mutación no se halló en ninguno de los dos padres, por lo que se trataría de una mutación de novo. Los pacientes con R453C y V822M habían sido trasplantados a los 43 y los 22 años, respectivamente.
Mutaciones en MYBPC3
Hallamos 18 mutaciones en MYBPC3 en 20 de los 120 (16%) pacientes (tabla 3). Sólo 6 (33%) de las 18 eran conocidas. Todas las mutaciones en MYBPC3 afectaban a residuos aminoacídicos conservados entre especies. Cinco eran cambios en la pauta de lectura de la proteína por inserción/deleción de nucleótidos (A328fs del G, Q404fs del C, G532fs del G, M844fs ins GA y R891fs ins G), una un codón de parada (G263X) y 13 cambios de un aminoácido por otro. La media de edad al diagnóstico en estos pacientes fue 42 años y 8 (40%) tenían antecedentes familiares de MCH y/o MSC (tabla 1). En 10 de los casos esporádicos pudimos estudiar a alguno de sus familiares, y hallamos a varios portadores asintomáticos (tabla 3).
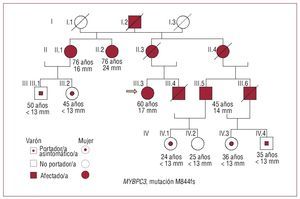
Un paciente era homocigoto para la mutación A627V, y fue el único con mutación en MYBPC3que había recibido un trasplante cardiaco. Dos familiares portadores de la mutación estaban clínicamente asintomáticos y sin hipertrofia. M844fs se identificó en un paciente del que 8 familiares también eran portadores, pero sólo 3 tenían síntomas clínicos de la enfermedad (fig. 2). Una paciente con la mutación R773H tenía también la mutación TNNT2-R278C, y se describe más adelante.

Fig. 2. Árbol de la familia con la mutación M844fs en el gen MYBPC3. El caso índice se indica con una flecha. Tres familiares de éste también eran portadores de la mutación, tenían síntomas clínicos de la enfermedad e hipertrofias de 14, 17 y 24 mm. Otros 5 portadores no tenían síntomas clínicos ni hipertrofia.
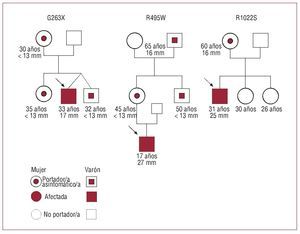
Tres pacientes jóvenes padecían disnea de esfuerzo durante la práctica deportiva (fig. 3). En estos casos se identificaron las mutaciones G263X, R495W y R1022S. El paciente G263X tenía 32 años y una hipertrofia de 19 mm; su madre, su hermana y un hermano gemelo eran también portadores de la mutación, pero estaban clínicamente asintomáticos y sin hipertrofia. El paciente R495W tenía 17 años y un septo de 27 mm; su madre, su tío y su abuelo también eran portadores, pero sólo éste tenía hipertrofia (18 mm) a la edad de 85 años. El paciente R1022S tenía 31 años y un septo de 25 mm; la madre también era portadora y no tenía síntomas clínicos, pero sí un septo de 18 mm.

Fig. 3. Familias de tres casos esporádicos con mutaciones en MYBPC3. Los casos índice se indican con una flecha. Los tres practicaban deporte regularmente y fueron diagnosticados tras presentar síntomas asociados al ejercicio físico. En las familias de los pacientes R495W y R1022S había otro portador sin síntomas de la enfermedad, pero con hipertrofia cardiaca.
Mutaciones en TNNT2, TNNI3 y TPM1
En 2 (1,66%) de los 120 pacientes hallamos las mutaciones R92Q y R278C en TNNT2 (tabla 2). El caso con R278C tenía también la mutación R733H en MYBPC3. En el gen TPM1 hallamos la mutación D175N en una paciente diagnosticada a los 41 años con una hipertrofia severa (32 mm) (tabla 2). Su hijo y su hermano tenían esta mutación y una hipertrofia de 27 y 20 mm.
Dobles portadores
Una mujer diagnosticada a los 49 años tenía dos mutaciones, R278C de TNNT2 y R733H de MYBPC3. Una hija y una nieta de 40 y 6 años eran dobles portadoras, pero se mantenían asintomáticas. Dos hijas de 47 y 42 años eran portadoras de la mutación R733H, y tampoco tenían síntomas clínicos ni hipertrofia. Una hermana de 52 años con la mutación R278C tenía síntomas leves y un septo de 13 mm.
Correlación genotipo-fenotipo
Se compararon las características clínicas y ecocardiográficas según el gen mutado (tabla 1). La media de edad al diagnóstico fue inferior en los casos con mutaciones en MYH7 comparados con MYBPC3, aunque la diferencia no fue significativa. Los pacientes sin ninguna mutación tenían una media de edad mayor que los pacientes con mutaciones, aunque las diferencias tampoco fueron significativas. La hipertrofia era de 21 ± 5 mm entre los pacientes MYH7, 22 ± 5 mm en los MYBPC3 y 19 ± 6 mm en los pacientes sin mutación. El 70% de los pacientes MYH7 tenían antecedentes de la enfermedad, comparados con el 40% de los MYBPC3y el 18% de los casos sin mutación.
Polimorfismos
Además de las mutaciones, se hallaron varios cambios no asociados a la enfermedad (polimorfismos) en los cinco genes. Todos estos cambios nucleotídicos fueron identificados también en los controles. En MYH7 se hallaron 27 polimorfismos, 23 de ellos en los exones y sólo uno implicaba un cambio de aminoácido (S1491C). En MYBPC3 se hallaron 24 polimorfismos, 11 exónicos, de los que 5 cambiaban el aminoácido: R17Q, S236G, R326Q, W382R y V896M. La información de la variación en estos genes puede ser solicitada a los autores en la dirección para correspondencia.
Discusión
Nuestro trabajo es el primero que analiza la secuencia completa de los cinco genes sarcoméricos más frecuentemente mutados en la MCH en una serie grande de pacientes españoles. Anteriormente se habían publicado series grandes para todos los exones de MYH7 o de un número reducido de casos para algunos exones de varios genes27-32.
El gen MYBPC3 fue el más mutado (el 16% de los casos), seguido de MYH7 (8%), y menos del 2% tenían mutaciones en los genes TNNT2 y TPM1. Hallamos una menor frecuencia de casos con mutaciones en comparación con las descritas por otros autores. La mayoría de esos estudios se han realizado en centros de referencia a los que se remite a pacientes con formas severas de MCH, en los que sería más probable hallar antecedentes familiares de la enfermedad. En nuestro estudio, sólo el 29% de los casos tenían antecedentes de MCH y/o MSC, mientras que otros estudios tenían hasta un 90% de formas familiares8,12. La frecuencia de mutaciones sarcoméricas sería mayor entre los pacientes con antecedentes familiares de la enfermedad, y el menor número de mutaciones identificadas en nuestro estudio podría deberse a una mayor frecuencia de casos esporádicos. Por otro lado, en el 43% de los pacientes con historia familiar de MCH no se hallaron mutaciones, lo que indica que otros genes podrían explicar la segregación familiar en estos casos.
La baja frecuencia de mutaciones en MYH7 (8%) ha sido descrita por otros, como Laredo et al30 en pacientes de Galicia. El 61% de los pacientes con mutaciones en MYBPC3 eran esporádicos, comparados con sólo un 30% de los MYH7. Ya había sido documentada3,8,12,15,33 una mayor penetrancia para las mutaciones en MYH7, lo que incrementaría la probabilidad de que la enfermedad se manifieste en los portadores en cada familia. Los pacientes con mutaciones en MYH7 manifestarían la enfermedad a edad más temprana, con mayor grado de hipertrofia, un fenotipo más maligno y un peor pronóstico8. Aunque hallamos una menor edad de manifestación de la enfermedad entre los pacientes MYH7,la diferencia con los MYBPC3 y los casos sin mutación no alcanzó significación estadística, probablemente debido al reducido número de pacientes con mutaciones. Tampoco se hallaron diferencias en el tamaño del septo interventricular entre los tres grupos. La baja frecuencia de mutaciones en los genes que codifican los filamentos finos TNNT2 y TPM1(< 2%) es similar a la descrita por otros grupos8.
La clasificación inicial de las mutaciones como «malignas» o «benignas» ha sido matizada por estudios más recientes, que han puesto de manifiesto la dificultad de estratificar el pronóstico para la mayoría de las mutaciones8,12-14. Dos de nuestros casos ilustran esta heterogeneidad clínica, incluso entre los portadores en una familia. Una mujer con dos mutaciones en TNNT2 y MYBPC3 había sido diagnosticada por angina y disnea a los 49 años y tenía una hipertrofia de 22 mm. Una hija y una nieta también eran dobles portadoras, pero estaban clínicamente asintomáticas y sin hipertrofia. Esto indica que la presencia de dos mutaciones sarcoméricas no estaría necesariamente asociada a una manifestación precoz y severa de la enfermedad. En otra familia con la mutación en MYBPC3 M844fs se identificaron 9 portadores, de los que sólo 3 tenían hipertrofia. Algunos portadores se mantenían asintomáticos a edad avanzada, pero 2 habían sufrido muerte súbita a edad < 50 años. Estos casos indican que las manifestaciones clínicas son consecuencia de factores de riesgo genéticos y no genéticos y, por lo tanto, la información genética de cada paciente no debería emplearse como único elemento para establecer el pronóstico. Sin embargo, las personas sin enfermedad cardiaca pero portadoras de mutaciones en genes sarcoméricos podrían manifestar síntomas más adelante, por lo que deberían someterse a evaluaciones periódicas para evitar los efectos adversos de la MCH.
Tres pacientes fueron diagnosticados ecográficamente tras padecer fatiga asociada al ejercicio físico. El estudio genético identificó tres mutaciones en MYBPC3 en las tres familias, en las que hallamos portadores asintomáticos. Estos tres casos indican que algunas mutaciones, particularmente en MYBPC3, podrían ser de baja penetrancia, pero el ejercicio físico aceleraría el desarrollo de los síntomas y la hipertrofia entre los portadores. Una MCH en deportistas sin antecedentes de la enfermedad podría indicar la presencia de alguna mutación sarcomérica, probablemente en MYBPC3.
Todas las mutaciones en MYH7 se traducían en cambios de un solo aminoácido, mientras que en MYBPC3 también había cambios en la pauta de lectura de la proteína. Además, los polimorfismos con cambio de aminoácido eran menos frecuentes en MYH7. Esto indica una presión selectiva en contra de las mutaciones que modifican varios aminoácidos de la cadena pesada de la betamiosina que podrían conllevar un riesgo elevado de muerte precoz, por lo que no se hallarían entre adultos con MCH8.
Finalmente, la mitad de las mutaciones en MYH7(5/10) y la mayoría de las halladas en MYBPC3(13/18) no habían sido descritas previamente33.
Sólo 2 de las 11 mutaciones halladas por Laredo et al30 (R663H y K1459N) se hallaron también en nuestros pacientes. Esto indica que los análisis directos de mutaciones ya conocidas tendrían escasa utilidad pues no identifican mutaciones en casos realmente portadores34. En éstos sería necesaria la secuenciación completa de los genes sarcoméricos para excluir de forma definitiva la presencia de alguna mutación.
Conclusiones
En conclusión, en un análisis de los cinco genes sarcoméricos más frecuentemente mutados en la MCH en una serie de 120 pacientes de Asturias y Cantabria, se hallaron mutaciones en el 26% de los casos. El gen MYBPC3 era el más mutado, seguido de MYH7, TNNT2 y TPM1. Más de la mitad de las mutaciones no habían sido descritas. No hallamos mutaciones en TNNI3. No había diferencias ni en la edad media al diagnóstico ni en el tamaño del septo interventricular entre los portadores en MYH7 y MYBPC3. Nuestro estudio ilustra la dificultad para definir el pronóstico en los portadores de mutaciones en estos genes.
AGRADECIMIENTOS
Queremos expresar nuestro agradecimiento a los pacientes y sus familiares.
ABREVIATURAS
MCH: miocardiopatía hipertrófica.
MSC: muerte súbita cardiaca.
Full English text available from: www.revespcardiol.org
Estudio financiado por proyecto FIS 06/0214, del Fondo de Investigaciones Sanitarias-Fondos FEDER Unión Europea (Investigador principal, Eliecer Coto García). Red de Investigación Renal-REDINREN (RD06/0016) del Instituto de Salud Carlos III. Eliecer Coto es beneficiario del Programa de Intensificación de la Actividad Investigadora del Instituto de Salud Carlos III.
Correspondencia: Dr. E. Coto.
Laboratorio de Genética Molecular. Hospital Universitario Central de Asturias (Maternidad).
33006 Oviedo. España.
Correo electrónico: eliecer.coto@sespa.princast.es
Recibido el 28 de abril de 2008.
Aceptado para su publicación el 12 de septiembre de 2008.