Introducción y objetivos
Estudios previos han demostrado que el losartán, antagonista de los receptores de tipo AT-1 de la angiotensina II (Ang II) podría bloquear al receptor del tromboxano A 2 (TXA 2) en la pared vascular. El objetivo del trabajo es estudiar el efecto del losarta´n sobre la activación de las plaquetas humanas.
Material y Métodos
Las plaquetas fueron obtenidas de 15 voluntarios sanos con edades comprendidas entre los 26 y 40 años. La activación plaquetaria fue medida por cambios en la transmisión de luz del plasma rico en plaquetas estimuladas por un análogo sintético del TXA 2 el U46619
. Resultados
El U46619 estimuló la agregación de las plaquetas, siendo significativamente inhibida por el losartán de manera dosis dependiente. Sólo dosis altas del EXP 3174, el metabolito hepático principal del losartán, consiguieron inhibir la activación plaquetaria inducida por el U46619. Captopril, inhibidor de la enzima convertidora de angiotensina, no fue efectivo en modificar la agregación plaquetaria inducida por el análogo del TXA 2.
Conclusión
Losartán disminuyó la agregación plaquetaria por un mecanismo dependiente de TXA 2 por un mecanismo independiente del bloqueo de los receptores del tipo A-1.
Palabras clave
Angiotensina
Plaquetas
Trombosis
Tromboxano
INTRODUCCIÓN
El péptido angiotensina II (Ang II) desempeña un importante papel en alteraciones cardíacas y vasculares. La estimulación del receptor AT-1 ha sido asociado con muchas de las acciones biológicas conocidas de la Ang II. El losartán es un potente antagonista de los receptores AT-1 no peptídico que produce una inhibición de la vasoconstricción inducida por la Ang II 1. El losartán disminuye la presión arterial en sujetos hipertensos, así como en modelos animales de hipertensión 2,3. El EXP 3174, el principal metabolito hepático del losartán, es aproximadamente de 10 a 15 veces más potente que el losartán en antagonizar el receptor AT-1 1.
La evaluación del losartán en el estudio ELITE ha demostrado que el tratamiento de pacientes con insuficiencia cardíaca con losartán causó una aparente mejoría en el índice de supervivencia, comparado con los tratados con captopril, un inhibidor de la enzima de conversión de la angiotensina I 4. Esta mejoría en la mortalidad en los pacientes tratados con losartán fue debida a una reducción en la muerte súbita cardíaca. Tanto la activación y la agregación plaquetaria, seguidas de trombosis, parecen contribuir al mecanismo de muerte súbita cardíaca 5.
El TXA 2 es un amplificador en la señal de la activación plaquetaria, siendo sintetizado y liberado en respuesta a una gran variedad de agonistas, como por ejemplo el adenosín difosfato 6-8. Li et al han demostrado recientemente que el losartán puede antagonizar el receptor de tromboxano A 2 (TXA 2)/prostanglandina H 2 en arteria coronaria de perro 9. Además, el losartán redujo de forma dosis-dependiente la hipertensión pulmonar inducida por una agonista del receptor de TXA 2 in vivo10.
Por tanto, basándonos en todos estos hallazgos, el objetivo del presente estudio fue analizar el efecto del losartán sobre la activación de plaquetas humanas.
MATERIALES Y MÉTODOS
Materiales
El [ 3H]-U46619 fue obtenido de Dupont NEN, Boston, MA. El losartán y EXP 3174 (metabolito hepático activo del losartán) fueron obtenidos por Meck, Sharp and Dohme, S.A. El resto de compuestos químicos, incluido el captopril, fueron obtenidos de Sigma Chemical Co. (St. Louis, Missouri, EE.UU.).
Preparación del plasma rico en plaquetas y agregación plaquetaria
El plasma rico en plaquetas (PRP) fue aislado de sangre periférica de 15 voluntarios sanos, recogido en 5% (v/v) de citrato-dextrosa y centrifugado a 1.000 rpm durante 10 min. Los voluntarios no habían recibido ningún fármaco en los 20 días antes de realizar los experimentos. El PRP fue recogido y el número de plaquetas fue ajustado con plasma pobre en plaquetas (PPP), obteniendo del mismo donante hasta 14 ¥ 10 7 células/ml de plasma. La activación plaquetaria fue evaluada en un lumiagregómetro (Aggrecorder de dos canales, Chrono-log Corporation Hovertown, Pennsylvania, EE.UU.) por cambios en la transmisión de luz 11,12. El PPP fue usado como control del 100% de transmisión de luz.
El PRP (500 ml) fue preincubado a 37 °C durante 3 min en el agregómetro con continuo movimiento (600 g) y posteriormente las plaquetas se estimularon con un agonista del TXA 2, el U46619 (5 ¥ 10 -6 mol/l).
Para estandarizar las medidas, sólo se usaron los valores de turbidez a los 6 min para los cálculos. Este período de tiempo corresponde al máximo valor de la primera onda de la agregación plaquetaria. Esta primera onda representa mejor la activación que la agregación plaquetaria y es parcialmente reversible. Losartán, EXP 3174 o captopril fueron añadidos a las plaquetas 5 min antes de que fueran estimuladas con el U46619. Se realizaron curvas dosis-respuesta de losartán y EXP3174, siendo la dosis mínima utilizada 5 ¥ 10 -9 mol/l y la máxima 5 ¥ 10 -5 mol/l. En el caso del captopril se utilizó una dosis de 5 ¥ 10 -5 mol/l.
En todos los casos, la medida basal comparativa fue realizada en presencia del disolvente de los fármacos, 0,8% de NaHCO 3 (bicarbonato sódico).
Ensayo de unión del receptor del TXA2
La unión del [ 3H]-U46619 (Dupont NEN, Boston, MA) a las plaquetas se realizó como describieron Kattelman et al 13. El PRP fue incubado con 1 mmol/l de ácido acetilsalicílico durante 30 min a 37 °C con el fin de inhibir la formación de TXA 2 endógeno. Entonces, las plaquetas fueron centrifugadas a 1.100 ¥ g durante 10 min y resuspendidas en tampón HEPES/Tyrode's (134 mmol/l de NaCl; 12 mmol/L NaHCO 3, 2,9 mmol/L KCl, 0,34 mmol/l de NaHPO 4; 1 mmol/l de MgCl 2; 10 mmol/l HEPES; 5 mmol/l de dextrosa; un 0,3% de BSA y un pH de 7,4) hasta una concentración final de 3 ¥ 10 8 plaquetas/ml. La suspensión de plaquetas se incubó con 4 nmol/l de [ 3H]-U46619 en presencia o ausencia de concentraciones crecientes de losartán y EXP 3174 durante 30 min a 37 °C.
La unión específica se determinó utilizando un exceso de 1.000 veces de U46619 no marcado. La reacción se paró mediante centrifugación a 7.000 ¥ g durante 10 min a 4 °C. El pellet fue lavado dos veces con tampón frío y transferido a un vial de centelleo para su recuento en contador de centelleo líquido (Beckman Instrumenst Inc., Fullerton, CA).
Método estadístico
Los resultados fueron expresados como la media ± error estándar de la media (EEM). Para determinar la significación estadística se realizó un test de ANOVA y corrección de Bonferroni de comparación múltiple o el test de la t de Student (apareado o no apareado). Un valor de p < 0,05 fue considerado estadísticamente significativo. RESULTADOS
Efecto del losartán sobre la activación plaquetaria
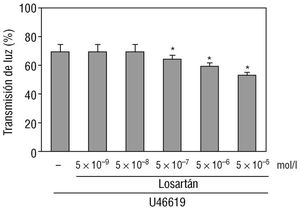
El porcentaje de transmisión de luz observado en las plaquetas humanas activadas con el agonista del TXA 2, el U46619 (5 ¥ 10 -6 mol/l) fue de 72 ± 4 (n = 5). El losartán redujo de forma significativa la activación plaquetaria inducida por el U46619 de forma dependiente de la dosis (fig. 1). Se observó una inhibición significativa de la activación de las plaquetas con una concentración de losartán de 5 ¥ 10 -7 mol/l (fig. 1). El máximo efecto del losartán se obtuvo a 5 ¥ 10 -5 mol/l, que fue la dosis máxima utilizada en estos estudios. Esta concentración fue usada para los siguientes experimentos.
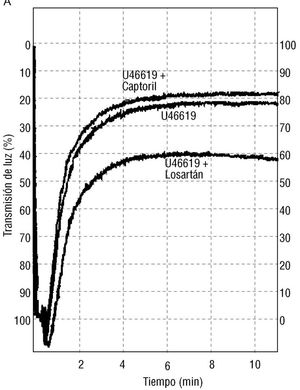
El dato representado en la figura 1 fue obtenido 6 min después del estímulo y corresponde a la máxima activación plaquetaria (fig. 2A). Sin embargo, las curvas de turbidimetría en presencia o ausencia del losartán fueron ya diferentes cuando fueron medidas 1 min después de comenzar los experimentos (fig. 2A). La adición de un inhibidor de la enzima de conversión, captopril (5 ¥ 10 -5 mol/l) no modificó la activación plaquetaria inducida por el U46619 (figs. 2A y B).
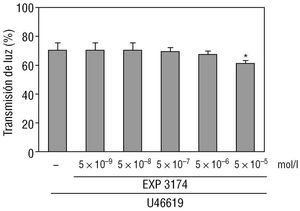
El metabolito hepático principal del losartán, EXP 3174, también disminuyó la activación de las plaquetas inducida por U46619 (fig. 3). Sin embargo, sólo la máxima dosis del EXP 3174 (5 ¥ 10 -5 mol/l) indujo una inhibición que alcanzó significación estadística (fig. 3). Ninguna otra dosis de EXP 3174 modificó de forma significativa la activación de las plaquetas inducida por U46619 (fig. 3).
La preincubación de las plaquetas con EXP 3174 (5 ¥ 10 -6 mol/l) no cambió el efecto de 5 ¥ 10 -6 mol/l de losartán sobre la estimulación de las plaquetas con el U46619 (porcentaje de transmisión de luz: losartán = 60 ± 1; EXP 3174 + losartán = 57 ± 2; n = 5; p = NS).
La activación espontánea de las plaquetas (porcentaje de transmisión de luz < 5%) no fue modificado por la incubación de PRP con 5 ¥ 10 -5 mol/l de losartán, 5 ¥ 10 -5 mol/l de EXP 3174 o 5 ¥ 10 -5 mol/l de captopril (porcentaje de transmisión de luz: losartán = 4 ± 2; EXP 3174 = 4 ± 1; captopril: 5 ± 1; n = 5; p = NS).
Efecto de la angiotensina II sobre la activación plaquetaria
No se observó ningún efecto de la Ang II (10 -7 mol/l) sobre la activación plaquetaria inducida por 5 ¥ 10 -5 mol/l de U46619 (porcentaje de transmisión de luz: 5 ¥ 10 -5 mol/l del U46619 = 85 ± 3; 5 ¥ 10 -5 mol/l de U46619 + 10 -7 mol/l de Ang II= 88 ± 2; n = 6; p = NS). Se realizaron también experimentos en presencia de dosis menores de U46619 (5 ¥ 10 -6 mol/l) con el fin de asegurarnos de que la falta de respuesta de las plaquetas a la Ang II exógena no fuera debida a que estuviéramos obteniendo una respuesta máxima con 5 ¥ 10 -5 mol/l de U46619 y, por tanto, no observaríamos potenciación de esa respuesta. La Ang II tampoco modificó la activación plaquetaria inducida por una dosis menor de U46619 (5 ¥ 10 -6 mol/l). En ausencia del agonista de TXA 2, la adición de Ang II a las plaquetas no indujo ningún tipo de modificación de la agregación espontanea (porcentaje de transmisión de luz < 5%).
Finalmente, analizamos si la falta de respuesta a la Ang II por las plaquetas se debía a una rápida degradación del péptido por la angiotensinasa. Para ello utilizamos un inhibidor de la angiotensinasa, el 1,10-0 fenantrolina. En presencia del inhibidor de la angiotensinasa, el 1,10-0 fenantrolina (5 ¥ 10 -6 mol/l), la Ang II (10 -7 mol/l) tampoco modificó la activación plaquetaria inducida por el U46619 (porcentaje de transmisión de luz: U46619 = 72 ± 4, U46619 + Ang II + 1,10-0 fenantrolina = 76 ± 3; n = 6; p = NS).
Interacción del losartán con el receptor del TXA2
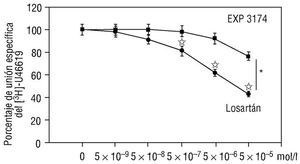
El desplazamiento de la unión del [ 3H]-U46619 a las plaquetas humanas por el losartán se expone en la figura 4. El losartán compitió de forma dosis-dependiente con el radioligando U46619, obteniéndose un 50% de inhibición de la unión (IC50) con 8 ¥ 10 -7 mol/l de losartán. El metabolito del losartán, EXP 3174, redujo de forma significativa la unión del [ 3H]-U46619 sólo al testar una dosis máxima de 5 ¥ 10 -5 mol/l (fig. 4). Por otro lado, el captopril (5 ¥ 10 -5 mol/l) no modificó la unión del [ 3H]-U46619 a las plaquetas (porcentaje de unión específica del [ 3H]-U46619 en presencia de 5 ¥ 10 -5 mol/l de captopril = 98 ± 1; n = 5; p = NS).

Fig. 1. En la gráfica de barras se observa el efecto de concentraciones crecientes de losartán sobre la activación plaquetaria inducida por un análogo del TXA2, el U46619 (5 ¥ 10-6 mol/l). La activación plaquetaria se representa como el porcentaje de transmisión de luz 6 min después de la adición de U46619. Los resultados se representan como la media ± error estándar de la media de 5 experimentos diferentes. *p < 0,05 con respecto al porcentaje de la transmisión de luz en ausencia de losartán.

Fig. 2. A: la gráfica es un trazado real de la agregación plaquetaria en respuesta al U46619 (5 ¥ 10-6 mol/l). Se representa también un experimento en presencia de un inhibidor de la enzima convertidora de angiotensina, el captopril (5 ¥ 10-5 mol/l), y del antagonista de los receptores AT-1 de angiotensina II, el losartán (5 ¥ 10-5 mol/l). B: en la gráfica de barras se exponen los resultados de 15 experimentos diferentes similares a los de la gráfica A. Los resultados están representados como la media ± error estándar de la media. *p < 0,005 con respecto al U46619 solo.

Fig. 3. En la gráfica de barras se observa el efecto de concentraciones crecientes del metabolito hepático activo del losartán, el EXP 3174, sobre la activación plaquetaria inducida por el U46619 (5 ¥ 10-6 mol/l). La activación plaquetaria fue representada como el porcentaje de transmisión de luz 6 minutos después de la adición de U46619. Los resultados están representados como la media ± error estándar de la media de 5 experimentos diferentes. *p < 0,05 con respecto al porcentaje de transmisión de luz en ausencia del EXP 3174.

Fig. 4. Esta gráfica representa los experimentos de unión del [3H]-U46619 y su desplazamiento por concentraciones crecientes de losartán y exp 3174 no marcados. La suspensión de plaquetas fue incubada con 4 nmol/l [3H]-U46619 en presencia o ausencia de concentraciones crecientes de losartán y EXP 3174. La unión específica se calculó usando concentraciones 1.000 veces superiores del U46619 no marcado. La figura representa la media ± error estándar de la media de 5 diferentes experimentos. *p < 0,05 con respecto al EXP 3174. Ip < 0,05 con respecto a la ausencia del fármaco correspondiente.
DISCUSIÓN
Los resultados de este trabajo aportan nuevas evidencias que demuestran efectos antiagregantes del antagonista de los receptores AT-1 de la Ang II, el losartán. Nuestros resultados apoyan la hipótesis de que el losartán actúa como un agente antiactivador plaquetario independientemente de sus efectos sobre los receptores de tipo AT-1 de la Ang II.
Losartán redujo de forma dosis dependiente la activación de las plaquetas humanas inducida por el análogo del TXA 2, U46619, disminuyendo además la unión del TXA 2 sobre su receptor plaquetario. Con el inhibidor de la enzima de conversión de la Ang I, captopril, no se observaron estos efectos.
Estudios previos han sugerido que el losartán podría interferir sobre el receptor del TXA 2 /PGH 2 en la pared vascular. Li et al habrían demostrado que el losartán inhibe la constricción inducida por TXA 2 de las arterias coronarias de perro 9. Además, el losartán reduce la hipertensión pulmonar inducida por un agonista de TXA 2, el U46619 10. Más recientemente, Corriu et al demostraron que el losartán puede actuar como un antagonista competitivo del receptor del TXA 2 en aortas de rata denudada de endotelio 14.
La capacidad del losartán para inhibir el receptor del TXA 2/PGH 2 no fue compartida por el EXP 3174, el metabolito activo in vivo del losartán. El EXP 3174 tiene una vida media en el plasma mayor que el losartán y es aproximadamente 10-15 veces más potente en bloquear el receptor AT-1 1. Sin embargo, nuestros resultados sugieren que no ocurre lo mismo para el caso del bloqueo del receptor de TXA 2 plaquetario. Otros efectos diferentes entre el losartán y el EXP 3174 han sido previamente publicados. En este sentido, los efectos uricosúricos del losartán en pacientes hipertensos esenciales e hipertensos con alteraciones renales han sido también atribuidos al losartán y no al EXP 3174, aunque los mecanismos por los que esto ocurre no están aún aclarados 15,16. Ambos, losartán y EXP 3174, contienen en su molécula un grupo imidazol, siendo la única diferencia estructural entre el metabolito y el losartán un grupo hidroxilo existente en el losartán que, por un mecanismo de oxidación, se convierte en un grupo carboxilo 1. Serían, por tanto, necesarios estudios con otros antagonistas AT-1 que contengan o no este tipo de radicales para poder dilucidar su importancia en la interacción con el receptor del TXA 2/PGH 2.
El estudio ELITE ha demostrado recientemente que el losartán redujo la muerte súbita cardíaca de una manera más efectiva que el captopril 4 y parece importante la contribución de las plaquetas activadas en la génesis de la muerte súbita cardíaca 5. En el presente estudio, el captopril no tuvo efecto sobre la activación plaquetaria, no así el losartán, que redujo la activación de las plaquetas abriendo una nueva hipótesis para poder explicar la aparente mejoría en la mortalidad de los pacientes tratados con losartán con respecto a los de captopril del estudio ELITE.
Sobre la base de los hallazgos presentados en este trabajo, la concentración de losartán requerida para inhibir la agregación plaquetaria (5 ¥ 10 -7 mol/l) fue muy alta comparada con la encontrada en el plasma humano tras la administración del fármaco (aproximadamente 5 ¥ 10 -9 mol/l), lo que sugeriría que con las dosis clínicas habituales del losartán no se alcanzaría un efecto relevante sobre las plaquetas. En nuestro estudio sólo se determinó el efecto directo del losartán sobre las plaquetas. Sin embargo, la trombosis es un evento multicelular en donde otras células, como los neutrófilos y el endotelio, regulan también la reactividad plaquetaria 17,18. En este sentido, se ha demostrado que la administración aguda de losartán incrementa el óxido nítrico que es, a su vez, un potente agente antiplaquetario 17,19. Por tanto, aunque sea de forma especulativa, la concentración de losartán que hipotéticamente inhibiría in vivo la activación plaquetaria podría ser significativamente más baja que en el estudio in vitro aquí presentado. En este sentido, a pesar de la aparente ausencia de un efecto directo del EXP 3174 y del captopril sobre el receptor de TXA 2 plaquetario, deberíamos tener en cuenta que la activación del receptor AT-1 expresado en la pared vascular por la Ang II podría favorecer la acumulación plaquetaria por un efecto vasoconstrictor independientemente de un efecto sobre el receptor de TXA 2. Futuros estudios realizados in vivo son necesarios para evaluar la relevancia práctica de estos hallazgos.
La adición de Ang II exógena no afectó significativamente a la activación plaquetaria. Además, la combinación de EXP 3174, que es un potente antagonista AT-1, con el losartán no modificó el efecto del losartán por sí mismo sobre la agregación de las plaquetas. Estos resultados sugieren que los efectos anteriormente mencionados sobre la activación plaquetaria fueron mediados por una acción no dependiente del receptor de tipo AT-1.
Los estudios de unión han demostrado previamente la presencia de receptores de tipo AT-1 en las plaquetas humanas 20,21. El efecto de la Ang II sobre la agregación plaquetaria continúa siendo controvertido. La potenciación de la activación plaquetaria por la Ang II fue publicado por Ding et al, pero como ocurre en este estudio, no fue confirmado por otros autores 22,23. De acuerdo con nuestros resultados, Burnier et al, usando el método de Fura-2, pudieron demostrar que no había ningún cambio significativo en el calcio citosólico después de la estimulación de plaquetas con Ang II, mientras que había un marcado incremento en el calcio citoplasmático después de la estimulación del receptor AT-1 por la Ang II en células del músculo liso vascular 23.
En resumen, los resultados del presente estudio sugieren que el losartán redujo la activación plaquetaria por un mecanismo dependiente de TXA 2. El metabolito in vivo del losartán, EXP 3174, demostró una menor potencia en inhibir el receptor del TXA 2 plaquetario. Este efecto no fue observado por el captopril. Desde un punto de vista clínico, no pensamos que el losartán pueda ser usado exclusivamente como un agente anti-TXA 2 ya que esta actividad no está presente en su metabolito, que tiene una vida media en plasma más larga. No obstante, esta actividad directa del losartán bloqueando el receptor de TXA 2 abre una nueva vía de investigación sobre los posibles mecanismos de actuación de estos fármacos. AGRADECIMIENTOS
Agradecemos a Begoña Ibarra su labor secretarial.
El péptido angiotensina II (Ang II) desempeña un importante papel en alteraciones cardíacas y vasculares. La estimulación del receptor AT-1 ha sido asociado con muchas de las acciones biológicas conocidas de la Ang II. El losartán es un potente antagonista de los receptores AT-1 no peptídico que produce una inhibición de la vasoconstricción inducida por la Ang II 1. El losartán disminuye la presión arterial en sujetos hipertensos, así como en modelos animales de hipertensión 2,3. El EXP 3174, el principal metabolito hepático del losartán, es aproximadamente de 10 a 15 veces más potente que el losartán en antagonizar el receptor AT-1 1.
La evaluación del losartán en el estudio ELITE ha demostrado que el tratamiento de pacientes con insuficiencia cardíaca con losartán causó una aparente mejoría en el índice de supervivencia, comparado con los tratados con captopril, un inhibidor de la enzima de conversión de la angiotensina I 4. Esta mejoría en la mortalidad en los pacientes tratados con losartán fue debida a una reducción en la muerte súbita cardíaca. Tanto la activación y la agregación plaquetaria, seguidas de trombosis, parecen contribuir al mecanismo de muerte súbita cardíaca 5.
El TXA 2 es un amplificador en la señal de la activación plaquetaria, siendo sintetizado y liberado en respuesta a una gran variedad de agonistas, como por ejemplo el adenosín difosfato 6-8. Li et al han demostrado recientemente que el losartán puede antagonizar el receptor de tromboxano A 2 (TXA 2)/prostanglandina H 2 en arteria coronaria de perro 9. Además, el losartán redujo de forma dosis-dependiente la hipertensión pulmonar inducida por una agonista del receptor de TXA 2 in vivo10.
Por tanto, basándonos en todos estos hallazgos, el objetivo del presente estudio fue analizar el efecto del losartán sobre la activación de plaquetas humanas.
MATERIALES Y MÉTODOS
Materiales
El [ 3H]-U46619 fue obtenido de Dupont NEN, Boston, MA. El losartán y EXP 3174 (metabolito hepático activo del losartán) fueron obtenidos por Meck, Sharp and Dohme, S.A. El resto de compuestos químicos, incluido el captopril, fueron obtenidos de Sigma Chemical Co. (St. Louis, Missouri, EE.UU.).
Preparación del plasma rico en plaquetas y agregación plaquetaria
El plasma rico en plaquetas (PRP) fue aislado de sangre periférica de 15 voluntarios sanos, recogido en 5% (v/v) de citrato-dextrosa y centrifugado a 1.000 rpm durante 10 min. Los voluntarios no habían recibido ningún fármaco en los 20 días antes de realizar los experimentos. El PRP fue recogido y el número de plaquetas fue ajustado con plasma pobre en plaquetas (PPP), obteniendo del mismo donante hasta 14 ¥ 10 7 células/ml de plasma. La activación plaquetaria fue evaluada en un lumiagregómetro (Aggrecorder de dos canales, Chrono-log Corporation Hovertown, Pennsylvania, EE.UU.) por cambios en la transmisión de luz 11,12. El PPP fue usado como control del 100% de transmisión de luz.
El PRP (500 ml) fue preincubado a 37 °C durante 3 min en el agregómetro con continuo movimiento (600 g) y posteriormente las plaquetas se estimularon con un agonista del TXA 2, el U46619 (5 ¥ 10 -6 mol/l).
Para estandarizar las medidas, sólo se usaron los valores de turbidez a los 6 min para los cálculos. Este período de tiempo corresponde al máximo valor de la primera onda de la agregación plaquetaria. Esta primera onda representa mejor la activación que la agregación plaquetaria y es parcialmente reversible. Losartán, EXP 3174 o captopril fueron añadidos a las plaquetas 5 min antes de que fueran estimuladas con el U46619. Se realizaron curvas dosis-respuesta de losartán y EXP3174, siendo la dosis mínima utilizada 5 ¥ 10 -9 mol/l y la máxima 5 ¥ 10 -5 mol/l. En el caso del captopril se utilizó una dosis de 5 ¥ 10 -5 mol/l.
En todos los casos, la medida basal comparativa fue realizada en presencia del disolvente de los fármacos, 0,8% de NaHCO 3 (bicarbonato sódico).
Ensayo de unión del receptor del TXA2
La unión del [ 3H]-U46619 (Dupont NEN, Boston, MA) a las plaquetas se realizó como describieron Kattelman et al 13. El PRP fue incubado con 1 mmol/l de ácido acetilsalicílico durante 30 min a 37 °C con el fin de inhibir la formación de TXA 2 endógeno. Entonces, las plaquetas fueron centrifugadas a 1.100 ¥ g durante 10 min y resuspendidas en tampón HEPES/Tyrode's (134 mmol/l de NaCl; 12 mmol/L NaHCO 3, 2,9 mmol/L KCl, 0,34 mmol/l de NaHPO 4; 1 mmol/l de MgCl 2; 10 mmol/l HEPES; 5 mmol/l de dextrosa; un 0,3% de BSA y un pH de 7,4) hasta una concentración final de 3 ¥ 10 8 plaquetas/ml. La suspensión de plaquetas se incubó con 4 nmol/l de [ 3H]-U46619 en presencia o ausencia de concentraciones crecientes de losartán y EXP 3174 durante 30 min a 37 °C.
La unión específica se determinó utilizando un exceso de 1.000 veces de U46619 no marcado. La reacción se paró mediante centrifugación a 7.000 ¥ g durante 10 min a 4 °C. El pellet fue lavado dos veces con tampón frío y transferido a un vial de centelleo para su recuento en contador de centelleo líquido (Beckman Instrumenst Inc., Fullerton, CA).
Método estadístico
Los resultados fueron expresados como la media ± error estándar de la media (EEM). Para determinar la significación estadística se realizó un test de ANOVA y corrección de Bonferroni de comparación múltiple o el test de la t de Student (apareado o no apareado). Un valor de p < 0,05 fue considerado estadísticamente significativo. RESULTADOS
Efecto del losartán sobre la activación plaquetaria
El porcentaje de transmisión de luz observado en las plaquetas humanas activadas con el agonista del TXA 2, el U46619 (5 ¥ 10 -6 mol/l) fue de 72 ± 4 (n = 5). El losartán redujo de forma significativa la activación plaquetaria inducida por el U46619 de forma dependiente de la dosis (fig. 1). Se observó una inhibición significativa de la activación de las plaquetas con una concentración de losartán de 5 ¥ 10 -7 mol/l (fig. 1). El máximo efecto del losartán se obtuvo a 5 ¥ 10 -5 mol/l, que fue la dosis máxima utilizada en estos estudios. Esta concentración fue usada para los siguientes experimentos.
El dato representado en la figura 1 fue obtenido 6 min después del estímulo y corresponde a la máxima activación plaquetaria (fig. 2A). Sin embargo, las curvas de turbidimetría en presencia o ausencia del losartán fueron ya diferentes cuando fueron medidas 1 min después de comenzar los experimentos (fig. 2A). La adición de un inhibidor de la enzima de conversión, captopril (5 ¥ 10 -5 mol/l) no modificó la activación plaquetaria inducida por el U46619 (figs. 2A y B).
El metabolito hepático principal del losartán, EXP 3174, también disminuyó la activación de las plaquetas inducida por U46619 (fig. 3). Sin embargo, sólo la máxima dosis del EXP 3174 (5 ¥ 10 -5 mol/l) indujo una inhibición que alcanzó significación estadística (fig. 3). Ninguna otra dosis de EXP 3174 modificó de forma significativa la activación de las plaquetas inducida por U46619 (fig. 3).
La preincubación de las plaquetas con EXP 3174 (5 ¥ 10 -6 mol/l) no cambió el efecto de 5 ¥ 10 -6 mol/l de losartán sobre la estimulación de las plaquetas con el U46619 (porcentaje de transmisión de luz: losartán = 60 ± 1; EXP 3174 + losartán = 57 ± 2; n = 5; p = NS).
La activación espontánea de las plaquetas (porcentaje de transmisión de luz < 5%) no fue modificado por la incubación de PRP con 5 ¥ 10 -5 mol/l de losartán, 5 ¥ 10 -5 mol/l de EXP 3174 o 5 ¥ 10 -5 mol/l de captopril (porcentaje de transmisión de luz: losartán = 4 ± 2; EXP 3174 = 4 ± 1; captopril: 5 ± 1; n = 5; p = NS).
Efecto de la angiotensina II sobre la activación plaquetaria
No se observó ningún efecto de la Ang II (10 -7 mol/l) sobre la activación plaquetaria inducida por 5 ¥ 10 -5 mol/l de U46619 (porcentaje de transmisión de luz: 5 ¥ 10 -5 mol/l del U46619 = 85 ± 3; 5 ¥ 10 -5 mol/l de U46619 + 10 -7 mol/l de Ang II= 88 ± 2; n = 6; p = NS). Se realizaron también experimentos en presencia de dosis menores de U46619 (5 ¥ 10 -6 mol/l) con el fin de asegurarnos de que la falta de respuesta de las plaquetas a la Ang II exógena no fuera debida a que estuviéramos obteniendo una respuesta máxima con 5 ¥ 10 -5 mol/l de U46619 y, por tanto, no observaríamos potenciación de esa respuesta. La Ang II tampoco modificó la activación plaquetaria inducida por una dosis menor de U46619 (5 ¥ 10 -6 mol/l). En ausencia del agonista de TXA 2, la adición de Ang II a las plaquetas no indujo ningún tipo de modificación de la agregación espontanea (porcentaje de transmisión de luz < 5%).
Finalmente, analizamos si la falta de respuesta a la Ang II por las plaquetas se debía a una rápida degradación del péptido por la angiotensinasa. Para ello utilizamos un inhibidor de la angiotensinasa, el 1,10-0 fenantrolina. En presencia del inhibidor de la angiotensinasa, el 1,10-0 fenantrolina (5 ¥ 10 -6 mol/l), la Ang II (10 -7 mol/l) tampoco modificó la activación plaquetaria inducida por el U46619 (porcentaje de transmisión de luz: U46619 = 72 ± 4, U46619 + Ang II + 1,10-0 fenantrolina = 76 ± 3; n = 6; p = NS).
Interacción del losartán con el receptor del TXA2
El desplazamiento de la unión del [ 3H]-U46619 a las plaquetas humanas por el losartán se expone en la figura 4. El losartán compitió de forma dosis-dependiente con el radioligando U46619, obteniéndose un 50% de inhibición de la unión (IC50) con 8 ¥ 10 -7 mol/l de losartán. El metabolito del losartán, EXP 3174, redujo de forma significativa la unión del [ 3H]-U46619 sólo al testar una dosis máxima de 5 ¥ 10 -5 mol/l (fig. 4). Por otro lado, el captopril (5 ¥ 10 -5 mol/l) no modificó la unión del [ 3H]-U46619 a las plaquetas (porcentaje de unión específica del [ 3H]-U46619 en presencia de 5 ¥ 10 -5 mol/l de captopril = 98 ± 1; n = 5; p = NS).

Fig. 1. En la gráfica de barras se observa el efecto de concentraciones crecientes de losartán sobre la activación plaquetaria inducida por un análogo del TXA2, el U46619 (5 ¥ 10-6 mol/l). La activación plaquetaria se representa como el porcentaje de transmisión de luz 6 min después de la adición de U46619. Los resultados se representan como la media ± error estándar de la media de 5 experimentos diferentes. *p < 0,05 con respecto al porcentaje de la transmisión de luz en ausencia de losartán.

Fig. 2. A: la gráfica es un trazado real de la agregación plaquetaria en respuesta al U46619 (5 ¥ 10-6 mol/l). Se representa también un experimento en presencia de un inhibidor de la enzima convertidora de angiotensina, el captopril (5 ¥ 10-5 mol/l), y del antagonista de los receptores AT-1 de angiotensina II, el losartán (5 ¥ 10-5 mol/l). B: en la gráfica de barras se exponen los resultados de 15 experimentos diferentes similares a los de la gráfica A. Los resultados están representados como la media ± error estándar de la media. *p < 0,005 con respecto al U46619 solo.

Fig. 3. En la gráfica de barras se observa el efecto de concentraciones crecientes del metabolito hepático activo del losartán, el EXP 3174, sobre la activación plaquetaria inducida por el U46619 (5 ¥ 10-6 mol/l). La activación plaquetaria fue representada como el porcentaje de transmisión de luz 6 minutos después de la adición de U46619. Los resultados están representados como la media ± error estándar de la media de 5 experimentos diferentes. *p < 0,05 con respecto al porcentaje de transmisión de luz en ausencia del EXP 3174.

Fig. 4. Esta gráfica representa los experimentos de unión del [3H]-U46619 y su desplazamiento por concentraciones crecientes de losartán y exp 3174 no marcados. La suspensión de plaquetas fue incubada con 4 nmol/l [3H]-U46619 en presencia o ausencia de concentraciones crecientes de losartán y EXP 3174. La unión específica se calculó usando concentraciones 1.000 veces superiores del U46619 no marcado. La figura representa la media ± error estándar de la media de 5 diferentes experimentos. *p < 0,05 con respecto al EXP 3174. Ip < 0,05 con respecto a la ausencia del fármaco correspondiente.
DISCUSIÓN
Los resultados de este trabajo aportan nuevas evidencias que demuestran efectos antiagregantes del antagonista de los receptores AT-1 de la Ang II, el losartán. Nuestros resultados apoyan la hipótesis de que el losartán actúa como un agente antiactivador plaquetario independientemente de sus efectos sobre los receptores de tipo AT-1 de la Ang II.
Losartán redujo de forma dosis dependiente la activación de las plaquetas humanas inducida por el análogo del TXA 2, U46619, disminuyendo además la unión del TXA 2 sobre su receptor plaquetario. Con el inhibidor de la enzima de conversión de la Ang I, captopril, no se observaron estos efectos.
Estudios previos han sugerido que el losartán podría interferir sobre el receptor del TXA 2 /PGH 2 en la pared vascular. Li et al habrían demostrado que el losartán inhibe la constricción inducida por TXA 2 de las arterias coronarias de perro 9. Además, el losartán reduce la hipertensión pulmonar inducida por un agonista de TXA 2, el U46619 10. Más recientemente, Corriu et al demostraron que el losartán puede actuar como un antagonista competitivo del receptor del TXA 2 en aortas de rata denudada de endotelio 14.
La capacidad del losartán para inhibir el receptor del TXA 2/PGH 2 no fue compartida por el EXP 3174, el metabolito activo in vivo del losartán. El EXP 3174 tiene una vida media en el plasma mayor que el losartán y es aproximadamente 10-15 veces más potente en bloquear el receptor AT-1 1. Sin embargo, nuestros resultados sugieren que no ocurre lo mismo para el caso del bloqueo del receptor de TXA 2 plaquetario. Otros efectos diferentes entre el losartán y el EXP 3174 han sido previamente publicados. En este sentido, los efectos uricosúricos del losartán en pacientes hipertensos esenciales e hipertensos con alteraciones renales han sido también atribuidos al losartán y no al EXP 3174, aunque los mecanismos por los que esto ocurre no están aún aclarados 15,16. Ambos, losartán y EXP 3174, contienen en su molécula un grupo imidazol, siendo la única diferencia estructural entre el metabolito y el losartán un grupo hidroxilo existente en el losartán que, por un mecanismo de oxidación, se convierte en un grupo carboxilo 1. Serían, por tanto, necesarios estudios con otros antagonistas AT-1 que contengan o no este tipo de radicales para poder dilucidar su importancia en la interacción con el receptor del TXA 2/PGH 2.
El estudio ELITE ha demostrado recientemente que el losartán redujo la muerte súbita cardíaca de una manera más efectiva que el captopril 4 y parece importante la contribución de las plaquetas activadas en la génesis de la muerte súbita cardíaca 5. En el presente estudio, el captopril no tuvo efecto sobre la activación plaquetaria, no así el losartán, que redujo la activación de las plaquetas abriendo una nueva hipótesis para poder explicar la aparente mejoría en la mortalidad de los pacientes tratados con losartán con respecto a los de captopril del estudio ELITE.
Sobre la base de los hallazgos presentados en este trabajo, la concentración de losartán requerida para inhibir la agregación plaquetaria (5 ¥ 10 -7 mol/l) fue muy alta comparada con la encontrada en el plasma humano tras la administración del fármaco (aproximadamente 5 ¥ 10 -9 mol/l), lo que sugeriría que con las dosis clínicas habituales del losartán no se alcanzaría un efecto relevante sobre las plaquetas. En nuestro estudio sólo se determinó el efecto directo del losartán sobre las plaquetas. Sin embargo, la trombosis es un evento multicelular en donde otras células, como los neutrófilos y el endotelio, regulan también la reactividad plaquetaria 17,18. En este sentido, se ha demostrado que la administración aguda de losartán incrementa el óxido nítrico que es, a su vez, un potente agente antiplaquetario 17,19. Por tanto, aunque sea de forma especulativa, la concentración de losartán que hipotéticamente inhibiría in vivo la activación plaquetaria podría ser significativamente más baja que en el estudio in vitro aquí presentado. En este sentido, a pesar de la aparente ausencia de un efecto directo del EXP 3174 y del captopril sobre el receptor de TXA 2 plaquetario, deberíamos tener en cuenta que la activación del receptor AT-1 expresado en la pared vascular por la Ang II podría favorecer la acumulación plaquetaria por un efecto vasoconstrictor independientemente de un efecto sobre el receptor de TXA 2. Futuros estudios realizados in vivo son necesarios para evaluar la relevancia práctica de estos hallazgos.
La adición de Ang II exógena no afectó significativamente a la activación plaquetaria. Además, la combinación de EXP 3174, que es un potente antagonista AT-1, con el losartán no modificó el efecto del losartán por sí mismo sobre la agregación de las plaquetas. Estos resultados sugieren que los efectos anteriormente mencionados sobre la activación plaquetaria fueron mediados por una acción no dependiente del receptor de tipo AT-1.
Los estudios de unión han demostrado previamente la presencia de receptores de tipo AT-1 en las plaquetas humanas 20,21. El efecto de la Ang II sobre la agregación plaquetaria continúa siendo controvertido. La potenciación de la activación plaquetaria por la Ang II fue publicado por Ding et al, pero como ocurre en este estudio, no fue confirmado por otros autores 22,23. De acuerdo con nuestros resultados, Burnier et al, usando el método de Fura-2, pudieron demostrar que no había ningún cambio significativo en el calcio citosólico después de la estimulación de plaquetas con Ang II, mientras que había un marcado incremento en el calcio citoplasmático después de la estimulación del receptor AT-1 por la Ang II en células del músculo liso vascular 23.
En resumen, los resultados del presente estudio sugieren que el losartán redujo la activación plaquetaria por un mecanismo dependiente de TXA 2. El metabolito in vivo del losartán, EXP 3174, demostró una menor potencia en inhibir el receptor del TXA 2 plaquetario. Este efecto no fue observado por el captopril. Desde un punto de vista clínico, no pensamos que el losartán pueda ser usado exclusivamente como un agente anti-TXA 2 ya que esta actividad no está presente en su metabolito, que tiene una vida media en plasma más larga. No obstante, esta actividad directa del losartán bloqueando el receptor de TXA 2 abre una nueva vía de investigación sobre los posibles mecanismos de actuación de estos fármacos. AGRADECIMIENTOS
Agradecemos a Begoña Ibarra su labor secretarial.
Bibliografía
[1]
Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev 1993; 45: 205-251.
[2]
Angiotensin receptor antagonists: focus on losartan. Lancet 1995; 346: 1403-1407.
[3]
Nonpeptide angiotensin II receptor antagonist. VIII. Characterization of functional antagonist displayed by DuP 753, an orally active antihypertensive agent. J Pharmacol Exp Ther 1990; 252: 719-725.
[4]
Randomised trial of losartan versus captopril in patients over 65 with heart failure (evaluation of Losartan in the Elderly Study, ELITE). Lancet 1997; 349: 747-752.
[5]
Interaction of platelets with the vessel wall in the pathophysiology of sudden cardiac death. Circulation 1986; 73: 224-226.
[6]
Thromboxanes: a new group of biologically active compounds derived from prostaglandin endoperoxides. Proc Natl Acad Sci 1975; 72: 2294-2298.
[7]
Platelet activation and inhibition in unstable coronary syndromes. Am J Cardiol 1997; 80: 17E-20E.
[8]
Trombosis y enfermedad coronaria: neutrófilos, óxido nítrico y aspirina. Rev Esp Cardiol 1998; 51: 171-177.
[9]
Nonpeptide angiotensin II antagonist losartan inhibits thromboxane A2-induced contractions in canine coronary arteries. J Pharmacol Exp Ther 1997; 281: 1065-1070.
[10]
Prevention of thromboxane A2 receptor-mediated pulmonary hypertension by a nonpeptide angiotensin II type 1 receptor antagonist. J Pharmacol Exp Ther 1994; 268: 747-752.
[11]
Comparison of in vitro effects of trifusal and acetylsalicilic acid on nitric oxide synthesis by human neutrophils. Eur J Pharmacol 1998; 343: 57-65.
[12]
Aspirin-stimulated nitric oxide production by neutrophils after acute myocardial ischemia in rabbits. Circulation 1996; 94: 83-87.
[13]
Characterization of U46619 binding in unactivated intact human platelets and determinations of binding site affinities of four TXA2/PGH2 receptor antagonists. Thromb Res 1986; 41: 471-481.
[14]
Effects of losartan on contractile responses of conductance and resistance arteries from rats. J Cardiovasc Pharmacol 1995; 26: 688-692.
[15]
Pharmacodynamic activity of intravenous E-3174, an angiotensin II antagonist, in patients with essential hypertension. Am J Hypertens 1994; 7: 1035-1040.
[16]
Angiotensin II receptor antagonists. Potential in elderly patients with cardiovascular disease. Drugs Aging 1997; 10: 421-434.
[17]
Effects of aspirin on platelets-neutrophil interactions. Role of nitric oxide and endothelin-1. Circulation 1995; 91: 2080-2088.
[18]
Thromboregulation: multicellular modulation of platelet reactivity in hemostasis and thrombosis. FASEB J 1993; 7: 516-522.
[19]
Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol Rev 1991; 43: 109-141.
[20]
Identification of AT-1 receptors on human platelets and decreased angiotensin II binding in hypertension. J Hypertension 1993; 11: 5230-5231.
[21]
Angiotensin II receptors on human platelets. Circ Res 1981; 51: 314-320.
[22]
Angiotensin II effects on platelet function. J Hypertens 1985; 3: S251-S253.
[23]
In vitro effects of DuP 753, a nonpeptide angiotensin II receptor antagonist, on human platelets and rats vascular smooth muscle cells. Am J Hypertension 1991; 4: 438-443.