
Bicuspid aortic valve (BAV) disorder is the most common congenital heart disease. The aim of this study was to describe the characteristics of 0- to 18-year olds with BAV in a population-based registry.
MethodsData from all pediatric patients were obtained from the Spanish registry for pediatric patients with bicuspid aortic valve (REVAB) (< 18 years). For data analysis, patients with BAV were divided into 2 groups by their features: isolated BAV and BAV with associated congenital heart disease.
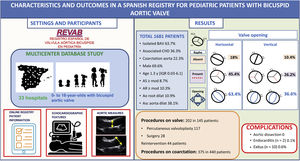
ResultsWe included 1681 patients from 33 hospitals. Males accounted for 69.6% (n = 1158). Valve morphology was horizontal in 63.4% (n = 1012) and pure (Sievers type 0) in 28.4% (n=469). Isolated BAV was present in 63.7% (n=1060), and concomitant left-sided obstructive lesions in 23.4% (n=390). Interventions were required in 8.6% (n=145).
ConclusionThese data represent the first large, population-based description of the clinical presentations and outcomes of patients enrolled in the Spanish registry for pediatric patients with bicuspid aortic valve.
Keywords
Bicuspid aortic valve (BAV) is the most common congenital cardiac abnormality, occurring in 1% to 2% of the general population.1–5 Although BAV by itself is considered a benign lesion, it should be considered a cluster of diseases involving different phenotypes, etiologies, and pathogenesis.5 This anomaly can be clinically silent, discovered incidentally on a transthoracic echocardiogram, or form part of a more complex congenital heart disease (a feature of Shone syndrome).6–9 Although BAV is often found in nonsyndromic individuals, it also can be a feature of connective tissue disorders (eg, Marfan, Loeys-Dietz, and vascular Ehlers-Danlos syndromes) and other syndromes, such as Turner and Beuren-Williams syndromes.10–12 BAV is characterized by a marked phenotypic heterogeneity of valve morphology; 3,13 and a widely accepted classification scheme has been described and is essential for meaningful discussions among authors.5
Children and adolescents with BAV are at risk of cardiovascular problems such as aortic stenosis, aortic insufficiency, and infective endocarditis, and the abnormality can be associated with aortic aneurysm and aortic dissection in adulthood.1,14,15
The objective of this study was to provide a population-based description of pediatric patients with BAV, including associated diseases, need for percutaneous or surgical interventions, and complications during childhood and adolescence.
METHODSStudy populationThis is the first analysis of the Spanish registry for pediatric patients with bicuspid aortic valve (REVAB), which is an open online registry under the auspices of the Spanish Society of Pediatric Cardiology and Congenital Heart Disease (SECPCC). This started in May 2016 for pediatric patients (< 18 years of age) with BAV. This is a descriptive and observational study. The study was approved by the ethics committees of all participating centers.
All pediatric cardiologists in the country were invited to participate. The registry was promoted during the official congress of the SECPCC and has been actively promulgated in all events endorsed by the SECPCC. In addition, a permanent link on the SECPCC website was provided,16 where all pediatric cardiologists who were interested could obtain information and enroll patients in the registry.
This is the first analysis including all patients registered from May 2016 to May 2021. A total of 1681 patients were introduced with the participation of 33 hospitals with different complexity levels (Appendix of the supplementary data). Some centers had databases and were able to include retrospective data, while others introduced prospective data only.
The following information was collected from each patient: a) demographic data such as age, sex, date of diagnosis, prenatal diagnosis, and family history of BAV (first-degree relatives diagnosed with BAV by echocardiography); b) associated congenital heart disease such left-sided obstructive lesions (LSOL), which include mitral stenosis, subaortic stenosis, aortic coarctation, and interrupted aortic arch; ventricular septal defect, tetralogy of Fallot, transposition of the great arteries and others; c). associated connective tissue diseases; d) classification of BAV by anatomical echocardiographic features: morphology of the aortic valve according to its opening pattern; e) the presence of complications derived from the BAV such as endocarditis, aortic dissection, or death; f) progress: visits performed at the outpatient clinic including weight, height, blood pressure, physical activity, diameter of the aortic root and ascending aorta, presence of aortic stenosis and/or regurgitation; g) procedures performed on the aortic valve or ascending aorta (surgical or percutaneous); and h) pharmacological treatment, if any, and dose (angiotensin converting-enzyme inhibitors, losartan, or beta-blockers). All this information was collected by the patients’ cardiologists.
In children and adolescents, the clinical phenotype is heterogeneous and is largely determined by age at presentation and concomitant heart defects. For a better understanding of our population, we divided the patients in 2 groups: those with isolated BAV, and those with BAV and associated congenital heart disease (CHD).
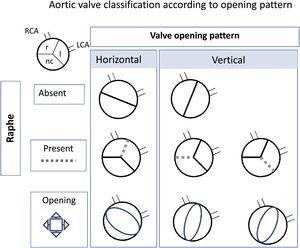
Echocardiographic featuresEchocardiographic examinations were conducted in all participants by the referring pediatric cardiologist. The valve opening pattern was classified according to the following: BAV was considered Sievers type 0 when 2 leaflets were present with no raphe, and Sievers type 1 when there was fusion between the 2 leaflets due to a presence of a raphe.17 For ease of interpretation, and because in pediatric patients it is difficult to see the raphes, we simplified the type of opening as horizontal when both coronary arteries arose from the same side of the opening and vertical when each coronary arose arose from a different side of the opening (figure 1).

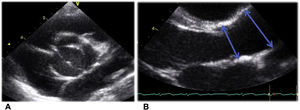
Aortic valve view and measurements were taken following the recommendations of the American Society of Echocardiography.18 The 2-dimensional parasternal long-axis view was used at the time of maximum expansion (mid-systole). The aortic root was measured at the level of the sinus of Valsalva and the ascending aorta was measured distal to the sinotubular junction as it crosses in front of the right pulmonary artery (figure 2).19
. The aortic root was measured at the level of the sinus of Valsalva and the ascending aorta was measured distal to the sinotubular junction as it crosses in front of the right pulmonary artery, as indicated by the arrows.")
Aortic valve view and measures following the recommendations of the American Society of Echocardiography21: A. 2-dimensional paraesternal short-axis view at the aortic valve level. B. 2-dimensional paraesternal long-axis view was used at the time of maximum expansion (mid-systole). The aortic root was measured at the level of the sinus of Valsalva and the ascending aorta was measured distal to the sinotubular junction as it crosses in front of the right pulmonary artery, as indicated by the arrows.
The hemodynamic status of the aortic valve was evaluated using color flow and spectral Doppler, significant aortic stenosis was defined when mean echocardiographic gradient was ≥ 20 mmHg, and significant aortic regurgitation was defined as ≥ moderate according to the echocardiographic findings: vena contracta> 0.3 cm, jet width> 25% of LVOT, pressure half-time <500 ms, and more than protodiastolic reversal aortic flow in the thoracic descending aorta.
Measurements of the aortic root and ascending aorta were adjusted to body surface area, and aortic dilatation was defined when the z-score was higher than + 2, based on Warren et al.20 regression equations.
Statistical analysisStatistical analysis was carried out by using the software package Stata V15.1 (StataCorp, United States).
Continuous variables are expressed as mean±standard deviation, median [interquartile range], or z-score. Categorical variables are expressed as the number of cases and proportions. Normality was evaluated in continuous variables using the Shapiro-Wilk test and was compared among groups using the Student t test and analysis of variance test. Categorical variables were compared using the chi-square test or Fisher exact test, as appropriate.
Survival analysis for mortality and procedures was performed using Kaplan-Meier curves. A P value <.05 was considered statistically significant.
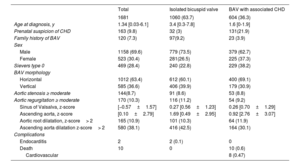
RESULTSDemographic featuresData were obtained from 1681 pediatric patients followed up at 33 institutions from all over Spain with different complexity levels. The patients’ demographic and clinical characteristics are shown in table 1.
Characteristics
| Total | Isolated bicuspid valve | BAV with associated CHD | |
|---|---|---|---|
| 1681 | 1060 (63.7) | 604 (36.3) | |
| Age at diagnosis, y | 1.34 [0.03-6.1] | 3.4 [0.3-7.8] | 1.6 [0-1.9] |
| Prenatal suspicion of CHD | 163 (9.8) | 32 (3) | 131(21.9) |
| Family history of BAV | 120 (7.3) | 97(9.2) | 23 (3.9) |
| Sex | |||
| Male | 1158 (69.6) | 779 (73.5) | 379 (62.7) |
| Female | 523 (30.4) | 281(26.5) | 225 (37.3) |
| Sievers type 0 | 469 (28.4) | 240 (22.8) | 229 (38.2) |
| BAV morphology | |||
| Horizontal | 1012 (63.4) | 612 (60.1) | 400 (69.1) |
| Vertical | 585 (36.6) | 406 (39.9) | 179 (30.9) |
| Aortic stenosis ≥ moderate | 144(8.7) | 91 (8.6) | 53 (8.8) |
| Aortic regurgitation ≥ moderate | 170 (10.3) | 116 (11.2) | 54 (9.2) |
| Sinus of Valsalva, z-score | [−0.57±1.57] | 0.27 [0.56±1.23] | 0.26 [0.70±1.29] |
| Ascending aorta, z-score | [0.10±2.79] | 1.69 [0.49±2.95] | 0.92 [2.76±3.07] |
| Aortic root dilatation, z-score> 2 | 165 (10.9) | 101 (10.3) | 64 (11.9) |
| Ascending aorta dilatation z-score> 2 | 580 (38.1) | 416 (42.5) | 164 (30.1) |
| Complications | |||
| Endocarditis | 2 | 2 (0.1) | 0 |
| Death | 10 | 0 | 10 (0.6) |
| Cardiovascular | 8 (0.47) | ||
BAV, bicuspid aortic valve; CHD, congenital heart disease; IQR, interquartile range.
Values are expressed as No. (%), mean±standard deviation, or median [interquartile range].
BAV was isolated in 63.7% of the patients (n=1060). The remaining 36.3% (621) had associated anomalies, which are detailed in table 2. Of note, 23.4% (n=390) had LSOL, including coarctation of the aorta (CoA) in 22.6% (n=375), interrupted aortic arch in 4% (n=6), subaortic stenosis in 2.8% (n=46), and mitral stenosis in 1.3%.21 Patients with connective tissue disease were included in this group.
Associated congenital heart disease and connective tissue diseases
| Associated lesions | N=621 |
|---|---|
| LSOL | 390 (62) |
| VSD | 206 (12.4) |
| ASD | 46 (2.8) |
| Persistent LSVC | 41 (2.5) |
| PDA | 52 (3.1) |
| AVSD | 3 (0.5) |
| Cor triatriatum | 3 (0.5) |
| Tetralogy of Fallot | 5 (0.3) |
| Transposition of great arteries | 6 (0.4) |
| Tuner syndrome | 4 (0.2) |
| CATCH22 syndrome | 4 (0.4) |
| Down syndrome | 3 (0.2) |
| Williams syndrome | 2 (0.1) |
| Noonan syndrome | 1 (0.1) |
| Alagille syndrome | 1 (0.1) |
| Connective tissue diseases | |
| Marfan syndrome | 2 (0.1) |
| Loeytz-Dietz syndrome | 4 (0.2) |
ASD, atrial septal defect; ASVD, atrioventricular septal defect; LSOL, left sided obstructive lesions; LSVC, left superior vena cava; PDA, patent ductus arteriosus; VSD, ventricular septal defect.
Data are expressed as no. (%).
Details on valve function and aortic disease (data available for 1564 patients [93%]) are shown in table 1. The median age at evaluation was 9.7 [5.5-14] years. Hypertension was present in 3.6% (n=37) and physical activity was classified as recreational in most of them (85%; n=747).
In patients with isolated BAV, up to 66.9% had some degree of valve dysfunction (aortic stenosis [AS] or aortic regurgitation [AR]), with AR being more frequent (AR 46.6% vs AS 20.3%) and classified mainly as trivial or mild with no clinical relevance. Vertical opening was associated with the severity of the aortic valve dysfunction compared with horizontal opening in this group, which had significant AS (12.1% vs 5.5%; P ≤ .05), and significant AR (16.7% vs 5.6%; P ≤ .05). Regarding aortic dilatation, significant AR was associated with both aortic root and ascending aorta dilatation (10.4% vs 8.6%; P=.002 and 42.9% vs 38.4; P ≤ .05). AS was associated only with ascending aorta dilatation (42% vs 33.1%, P ≤ .05). No association was found between aortic dilatation and the valve opening pattern in the isolated BAV group.
In patients with BAV and associated CHD, 52.7% of patients had some degree of valve dysfunction (AS or AR), with AR being more frequent (AR 30.4% vs AS 22.3%) and classified mainly as trivial or mild with no clinical relevance.
Vertical opening was associated with the severity of the aortic valve dysfunction when compared with horizontal opening in patients with BAV and associated CHD. AR was significant in this group (16.5% vs 5.6%; P ≤ .05). No association was found with AS. Regarding aortic dilatation, significant AR was associated with both aortic root and ascending aorta dilatation (12.1% vs 10.2%; P=.002 and 30.5% vs 25.5%; P ≤ .0001). No association between AS and aortic dilatation was found in this group.
Patients with CoA had less dilatation of the aortic root (8.1% vs 11.6%; P=.06) and ascending aorta (23.8% vs 42.2%; P <.0001) than patients without CoA.
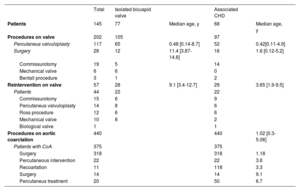
ProceduresA total of 490 patients (29.1%) underwent interventions related to BAV, most of them secondary to CoA repair (68%). In all, 202 BAV-related procedures were performed in 145 patients (9.1%) and 44 patients (2.5%) required reintervention (table 3).
Procedures
| Total | Isolated bicuspid valve | Associated CHD | |||
|---|---|---|---|---|---|
| Patients | 145 | 77 | Median age, y | 68 | Median age, y |
| Procedures on valve | 202 | 105 | 97 | ||
| Percutaneus valvuloplasty | 117 | 65 | 0.48 [0.14-8.7] | 52 | 0.42[0.11-4.9] |
| Surgery | 28 | 12 | 11.4 [3.87-14.8] | 16 | 1.6 [0.12-5.2] |
| Commissurotomy | 19 | 5 | 14 | ||
| Mechanical valve | 6 | 6 | 0 | ||
| Bentall procedure | 3 | 1 | 2 | ||
| Reintervention on valve | 57 | 28 | 9.1 [3.4-12.7] | 29 | 3.65 [1.9-9.5] |
| Patients | 44 | 22 | 22 | ||
| Commissurotomy | 15 | 6 | 9 | ||
| Percutaneus valvuloplasty | 14 | 8 | 6 | ||
| Ross procedure | 12 | 6 | 6 | ||
| Mechanical valve | 10 | 8 | 2 | ||
| Biological valve | 1 | 1 | |||
| Procedures on aortic coarctation | 440 | 440 | 1.02 [0.3-5.06] | ||
| Patients with CoA | 375 | 375 | |||
| Surgery | 318 | 318 | 1.18 | ||
| Percutaneous intervention | 22 | 22 | 3.6 | ||
| Recoartation | 11 | 118 | 3.3 | ||
| Surgery | 14 | 14 | 9.1 | ||
| Percutaneus treatment | 20 | 50 | 6.7 |
CHD, associated congenital heart disease; CoA, coartaction of aorta.
Values are expressed as No. (%), mean ± standard deviation, or median [interquartile range]
Infective endocarditis was reported in 2 (0.1%) patients. The first was a 3-year-old with a history of percutaneous treatment for BAV at the age of 2 months. The second was a foreign patient who had an ischemic stroke at the middle cerebral artery secondary to aortal infective endocarditis in the first year of life; surgical valvulotomy was performed and BAV was described as previously unknown. This patient is currently under follow-up in Spain. No deaths occurred in this cohort.
Exitus occurred only in the group of patients with BAV and associated CHD. There were 10 (0.6%) deaths at a mean age of 2.87 years (range, birth to 13.75 years). The cause of death was related to Shone syndrome in 5 patients, 2 died in the immediate postoperative period, 1 died at the age of 2 months due to pulmonary hypertension after aortic coarctation repair, 1 died after biventricular correction was attempted, and 1 associated CHARGE sequence (coloboma of the eye, heart defects, atresia of the nasal choanae, growth retardation and/or developmental delay, genital and/or urinary abnormalities, and ear abnormalities and deafness) and it was decided to limit therapeutic effort on the first day of life. One patient died at the age of 1 month after aortic coarctation repair with balloon angioplasty; in this patient the initial procedure was successful, but the patient was lost to follow-up and was readmitted 1 month later in cardiorespiratory arrest; imaging test and autopsy were refused by the family.
A total of 3 patients died due to coronary anomalies: hypoplasia of the coronary arteries and coronary fistulas were documented, respectively. One patient with associated CHARGE sequence had multiple episodes of sepsis, which was the main cause of death.
Overall, 157 (9.3%) patients had any event, including need for percutaneous treatment, surgery, endocarditis, and/or death (figure 1). No complications related to aortic dilation occurred in this cohort.
Pharmacological treatmentOnly 112 patients (6.8%) were under drug therapy (41% angiotensin converting enzyme inhibitors, 30% angiotensin II receptor antagonists, 27% beta-blockers).
DISCUSSIONTo the best of our knowledge this is the first open, large, and broad registry of pediatric patients with BAV and it is representative of the Spanish pediatric population. In this first analysis of the whole cohort, we describe the characteristics, valve morphology, associated diseases, interventions, and complications in 1681 pediatric patients diagnosed with BAV (figure 3). Due to the heterogeneity of BAV, we divided this cohort in 2 groups: isolated BAV and BAV with associated CHD.

Central illustration. AS, aortic stenosis; Asc aorta dilat, ascending aortic dilatation; Associated CHD, associated congenital heart disease; Ao root dilat, aortic root dilatation; AR, aortic regurgitation; BAV, bicuspid aortic valve; LCA, left coronary artery; nc, noncoronary; RCA, right coronary artery.
BAV was more frequent in males,21,22 and was mainly an isolated pathology. Patients with BAV and associated CHD were younger at diagnosis and there was greater prenatal suspicion of CHD than in patients with isolated BAV, probably due to the severity of the associated lesions.
Previous studies have demonstrated familial clustering of BAV.7,23,24 In our cohort, patients did not undergo prior family screening; we found that up to 13% patients with BAV (9.2% with isolated BAV and 3.9% with BAV and associated CHD) had familial clustering of BAV. This finding is similar to those of other series, which have reported that up to 10% of patients with BAV have first-degree-relatives with a history of left heart abnormalities.7,25,26 Currently, the American guidelines recommend echocardiographic screening in all first-degree relatives of patients with BAV.25
Associated congenital heart diseaseMore than one-third of pediatric patients with BAV had a concomitant cardiac anomaly, the most frequent being LSOL; this is compatible with the theory proposing that BAV with associated CHD is the mildest spectrum of hypoplastic left heart syndrome.27
The association between BAV with associated CHD and LSOL is widely known, with CoA being the most common concomitant defect and its prevalence is likely to be around 30% to 50%.6,14,27,28 In our data, the incidence of the CoA was 22.6%.
Anatomical echocardiographic featuresMost frequently, a BAV fused type with horizontal opening was identified in both groups, and vertical type was associated with a greater risk of valve dysfunction and interventions. These findings are broadly described in literature.10,20,28,29 Recently, a BAV consensus redefined valve classification.26 For the present study, we used the Sievers classification because it was designed prior to the consensus published by Michelena et al.,26 although we omitted cusp fusion as it is usually difficult to assess by echocardiography and this was a broad-based registry with a mix of expertise on this topic. In addition, similar classifications were previously used by others.27,30
Valve function and aortopathyThe most common clinical phenotype was an asymptomatic patient with no significant AS or AR. This finding is in line with those of other studies; 6,29 corroborating the notion that BAV may be a silent condition in children and adolescents. However, most patients had some degree of valve malfunction, mainly AR. The vertical opening pattern was associated with both significant AS and AR, confirming the findings previously described in other cohorts in which R-N (right-noncoronary) cusp fusion vertical opening is associated with future complications and worse prognosis.10,27,29,
Ascending aorta dilatation was relatively common in our cohort and was more frequent than aortic root dilatation. Dilatation of the ascending aorta was associated with AR and AS in both the isolated and concomitant CHD groups, and with a vertical opening pattern in the latter. In contrast, dilatation of the aortic root was only associated with AR. Several studies have reported a similar result,5,15,20,29,31–33 but to date, there has been no detailed analysis inferring isolated vs concomitant heart disease in the pediatric population.
In our cohort, pediatric patients with CoA were associated with a smaller ascending aorta. This is unsurprising as patients with CoA fall on the spectrum of hypoplastic left heart syndrome with varying severity, and hence they may represent a subgroup with a different pathophysiology and progress not yet clearly established in childhood and adolescence. This association has been previously described by other authors in pediatric cohorts,6,29 but contrasts with the results of studies in adults in which some authors reported that CoA is an even greater risk factor for ascending aorta complications in patients with BAV than in patients with isolated BAV.34 There is a need for studies that assess whether other risk factors acquired over the years (obesity, systemic arterial hypertension, etc) influence the progress of these patients.
ProceduresIn our cohort, almost 1 out of 10 patients (8.6%) underwent at least 1 intervention with a total of 202 procedures on the aortic valve and 42 reinterventions. Fernandes et al.27 found that percutaneous or surgical intervention during childhood and adolescence was needed in almost 1 out of 5 patients with BAV (about 15%). This difference may rely on the study population, as the study by Fernandes et al.35 was performed in a single tertiary center, with a referral bias. Nevertheless, it is known that BAV is associated with significant medical and surgical morbidity throughout life. According by Hill et al.,36 10-year freedom from AV replacement was 76% and valve reintervention was 46%. Interestingly, aortic procedures were similar in both the isolated and CHD groups. In both groups, AS is the main indication for any intervention on BAV. Significant AR (moderate or more) in valves without prior intervention is uncommon in childhood and adolescence.6,29
ComplicationsClassical complications associated with BAV were low in the pediatric population, including infective endocarditis, aortic dissection, and death.
Although native aortic valve endocarditis is rare in childhood and adolescence, patients with BAV have a significantly higher risk than the general population and the risk is even higher in those with previous intervention on the aortic valve.37 In our cohort, there were only 2 episodes of infective endocarditis in patients with isolated BAV.
Exitus was reported in only 10 patients, all of them with BAV and associated CHD with different spectrums of severity. There were no aortic dissections.
LimitationsThis study has some limitations, such as its mixed retrospective-prospective nature and voluntary reporting. Additionally, patients were included on the basis of echocardiographic data from hospitals with different complexity levels and gathered by pediatric cardiologists with variable experience, and consequently the measures may differ between operators, leading to relevant interobserver variability that may have influenced the results. Formal screening for first-degree relatives was not performed for this study; therefore, the percentage of family members with BAV may be underdiagnosed.
CONCLUSIONSBAV in childhood and adolescence is characterized by significant heterogeneity of its valvular and aortic phenotypic expressions, associated cardiac defects, and complications. Although BAV-related complications are unusual before adulthood, pediatric patients may show early onset valvular dysfunction, need for aortic procedures, and dilatation of the aorta. A long-term prospective and longitudinal community-based study is needed to further characterize the natural history of BAV beyond childhood and adolescence.
FUNDINGJuan V. Comas Grant of the Spanish Society of Pediatric Cardiology and Congenital Heart Disease.
AUTHORS’ CONTRIBUTIONSAll authors are responsible for the data contained in this manuscript. The authors confirm contribution to the manuscript as follows: study conception and design: A. Sabaté-Rotés; data collection: all authors; analysis and interpretation of results: A. Sabaté-Rotés, C. Juzga-Corrales; draft manuscript preparation: A. Sabaté-Rotés, C. Juzga-Corrales. All authors reviewed the results and approved the final version of the manuscript.
CONFLICTS OF INTERESTThe authors declare no conflicts of interest.
- •
Bicuspid aortic valve is the most common congenital cardiac abnormality, occurring in 1% to 2% of the general population. This anomaly can be clinically silent, discovered incidentally on a transthoracic echocardiogram, or form part of a more complex congenital heart disease. Pediatric patients with bicuspid aortic valve are at risk of cardiovascular problems such as aortic stenosis and aortic insufficiency, and the anomaly can be associated with aortic aneurysm and aortic dissection in adulthood.
- •
This is the first large and broad registry for pediatric patients with bicuspid aortic valve in Spain. In this pediatric population, there was a low prevalence of the classical complications associated with bicuspid aortic valve.
This work has been carried outunder the doctoral program of Paediatrics, Obstetrics and Gynaecology of the Universitat Autònoma de Barcelona. The statistical analysis was performed by the Statistics and Bioinformatics Unit of Vall d'Hebron Institute of Research (VHIR).
The authors warrant that the following investigators are responsible for the data contained in this manuscript:
- 1.
Hospital Universitari Vall d’Hebron, Barcelona, Spain: Carolina Juzga-Corrales and Anna Sabaté-Rotés.
- 2.
Hospital Clínico Universitario Lozano Blesa y Hospital Universitario Miguel Servet, Zaragoza, Spain: Ariadna Ayerza Casas.
- 3.
Hospital Universitari Dr. Josep Trueta, Girona, Spain: Marc Figueras Coll.
- 4.
Hospital Universitari Son Espases; Palma de Mallorca, Spain: Silvia Escribà Bori.
- 5.
Complejo Asistencial Universitario de Salamanca, Salamanca, Spain: Beatriz Plata Izquierdo.
- 6.
Hospital Universitario Sant Joan de Reus, Reus, Spain: Rosa Collell.
- 7.
Hospital General Universitario de Ciudad Real, Ciudad Real, Spain: María Aránzazu González Marín.
- 8.
Hospital HM Nens, Barcelona, Spain: José Manuel Siurana.
- 9.
Hospital General Universitario Santa Lucia, Cartagena, Spain: Moisés Sorli.
- 10.
Hospital Universitario 12 de Octubre, Madrid, Spain: Leticia Albert de la Torre.
- 11.
Hospital Universitari Parc Taulí, Barcelona, Spain: Silvia Teodoro Marín.
- 12.
Hospital HM Montepríncipe, Madrid, Spain: Mónica Rodríguez.
- 13.
Hospital Virgen de la Salud, Toledo, Spain: Olga Domínguez García.
- 14.
Hospital Clínico Universitario de Valladolid, Valladolid, Spain: Sara Rellán.
- 15.
Hospital Universitario Virgen del Rocío, Sevilla, Spain: Begoña Manso.
- 16.
Complejo Hospital Universitario Santiago de Compostela, Santiago de Compostela, A Coruña, Spain: Bernardo López Abel.
- 17.
Hospital de la Santa Creui I Sant Pau, Barcelona, Spain: Roser Álvarez Pérez.
- 18.
Hospital de Mérida, Mérida, Spain: Manuel Portillo Márquez.
- 19.
Hospital Universitario Donostia, San Sebastián, Spain: Erika Rezola.
- 20.
Hospital Universitario Río Hortega, Valladolid, Spain: Fernando Centeno Malfaz.
- 21.
Hospital Universitario Infanta Leonor, Madrid, Spain: Ruth Solana Gracia.
- 22.
Hospital Universitario Príncipe de Asturias, Madrid, Spain: Henar Rojo Sombrero.
- 23.
Complejo Asistencial Universitario de Palencia, Palencia, Spain: María Teresa Cantero Tejedor.
- 24.
Hospital San Pedro, Logroño, Spain: Bibiana Riaño.
- 25.
Hospital Universitario Reina Sofía, Córdoba, Spain: María Ángeles Tejero Hernández.
- 26.
Hospital General de Segovia, Segovia, Spain: Marisol Jiménez Casso.
- 27.
Hospital Universitari General de Cataluyna, Barcelona, Spain: Ana María Pérez Pardo.
- 28.
Hospital Lluis Alcanyis de Játiva, Valencia, Spain: Ana Moriano Gutiérrez.
- 29.
Hospital Nuestra Señora de Sonsoles, Ávila, Spain: Manuel Marrero Calvo.
- 30.
Hospital Universitario Infanta Elena, Madrid; Spain: María Teresa Fernández.
- 31.
Hospital Universitario de Jerez, Jerez de la Frontera, Spain: Carlos Salido Peracuala.
- 32.
Hospital Costa del Sol, Marbella, Spain: María José Bravo.
- 33.
Hospital Sanitas la Zarzuela, Madrid, Spain: Federico Gutiérrez-Larraya.
Supplementary data associated with this article can be found in the online version, at https://doi.org/10.1016/j.rec.2023.03.004