La disección aórtica tipo A es una patología de elevada mortalidad que requiere un diagnóstico y un tratamiento quirúrgico precoces. Este carácter emergente se debe a su alta capacidad para desarrollar de forma rápida complicaciones graves como insuficiencia aórtica grave, taponamiento cardiaco, ruptura aórtica e incluso la muerte, la cual ocurre en un alto porcentaje de casos.
Se sabe que una variedad de factores de riesgo contribuyen a la rigidez aórtica facilitando tanto el desarrollo de aneurismas como de disecciones, siendo la hipertensión arterial uno de los más importantes1. El engrosamiento intimal fibroso y el aumento de proteoglicanos y macrófagos/histiocitos CD68 positivos se observan habitualmente en el envejecimiento de las aortas. Estos hallazgos se relacionan con la ateroesclerosis1. Por otro lado, hay varios síndromes genéticos que se asocian con aneurismas de aorta ascendente y disecciones. Los más frecuentes y conocidos son el síndrome de Marfan y la válvula aórtica bicúspide, pero otros como el síndrome de Loeys-Dietz y el síndrome de Turner son también causas relativamente comunes. Estos síndromes comparten algunas características histopatológicas, principalmente la degeneración quística de la media1. Una alteración final común de estos síndromes es una regulación positiva de la actividad del factor de crecimiento transformador beta en la aorta ascendente2.
Más recientemente, se ha descrito la entidad conocida como aneurismas y disecciones aórticas familiares. Esta entidad presenta pocas características físicas evidentes y se ha asociado con mutaciones en múltiples genes, incluyendo la alfa-actina del músculo liso, el factor de crecimiento transformador beta y la cadena pesada de miosina 113. Para su diagnóstico se requiere: a) dilatación aórtica o disección tipo A o tipo B antes de los 60 años de edad, en ausencia de otros síndromes de tejido conectivo, hipertensión o enfermedad ateroesclerótica, y b) evidencia patológica de necrosis quística de la media aórtica o antecedentes familiares positivos (aneurisma o disección aórtica, muerte súbita inexplicada o mutación genética conocida)3.
Se espera que sean hereditarias hasta un 40% de las disecciones de aorta torácica; por lo que el estudio de un genotipo subyacente específico podría ayudar a confirmar el diagnóstico, llevar a cabo una vigilancia adecuada, permitir un tratamiento médico y quirúrgico individualizado y realizar un cribado de otros miembros de la familia con el objetivo de reducir la morbimortalidad4. Esto ha motivado que la American College of Cardiology Foundation y la American Heart Association, recomienden que la mutación genética subyacente deber dictar el momento de la reparación aórtica5.
Por lo tanto, dadas las implicaciones familiares que puede tener el hecho de reconocer dichas entidades, es fundamental llegar a un diagnóstico etiológico. Pero ¿llegamos siempre hasta el final?
Para responder a esta pregunta se diseñó un estudio cuyo objetivo era evaluar en cuántos de los pacientes diagnosticados de disección tipo A en nuestro centro entre los años 2000-2016 se realizó un diagnóstico etiológico final.
Se trata de un estudio retrospectivo en el que se revisaron todos los casos diagnosticados de disección tipo A en ese intervalo de tiempo. Se revisó la historia de los fallecimientos y la presencia o no de autopsia con el diagnóstico. En los supervivientes se recogió igualmente si había diagnóstico etiológico final.
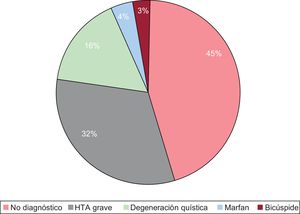
Se analizaron los datos de 75 pacientes, 47 de los cuales eran varones (63%), con una edad media al diagnóstico (63,09±13,8) y un seguimiento medio de 76±49 meses. Entre los factores de riesgo se contemplaba: hipertensión arterial (53,3%), diabetes mellitus (8%), dislipemia (15%) y ser fumador (12%). La tasa de fallecimiento intraoperatorio fue del 6,7% (5 pacientes) y la de fallecimiento intrahospitalario del 25,3% (19 pacientes). Se obtuvo necropsia en 5 de los pacientes fallecidos (6,7%). Durante el seguimiento hubo 6 fallecimientos (8%) y 11 pacientes perdidos (14,7%). Dos de los pacientes presentaban historia familiar y se había realizado estudio genético en 4 (5,3%). En el diagnóstico etiológico se obtuvo: síndrome de Marfan, 4% (3 pacientes); bicúspides, 2,7% (2 pacientes); degeneración quística descrita en 12 pacientes (16%), e hipertensión arterial grave en 24 (31,8%). En 34 pacientes (45,2%) no se estableció un diagnóstico etiológico final definitivo, si bien en 16 pacientes (21,2%) se hallaron cambios degenerativos en la pared aórtica (figura).

Con estos datos se llegó a la conclusión de que en un porcentaje significativo de casos de disección tipo A no se alcanzó un diagnóstico etiológico, siendo este dato fundamental para el consejo familiar. Las entidades sindrómicas son fácilmente diagnosticables, no así entidades como los aneurismas y las disecciones familiares que requieren cribado familiar y que se asocian con degeneración quística de la media. Por ello, tanto la historia familiar como el estudio histológico de la pared aórtica enferma, así como el estudio genético si está disponible, son claves en esta patología. Igualmente, es también importante el seguimiento de los pacientes diagnosticados de disección de aorta por equipos multidisciplinares capaces de mejorar la morbimortalidad tanto de los pacientes como, en algunos casos, de sus familiares.