

El papel de la genética en el diagnóstico y la personalización de los tratamientos de las aortopatías, es cada vez mayor. En este estudio se analizó la prevalencia de variantes genéticas en pacientes con síndrome aórtico agudo (SAA) admitidos consecutivamente en una unidad de cuidados intensivos y se evaluó su utilidad clínica.
MétodosMediante secuenciación masiva, se analizó 42 genes asociados a aortopatías en pacientes con SAA no sindrómico. Las variantes identificadas se segregaron mediante secuenciación Sanger en los familiares disponibles. Además, se estudió la relación entre los resultados genéticos y algunas características clínicas mediante la aplicación de los test exactos de Fisher y de Fisher-Freeman-Halton.
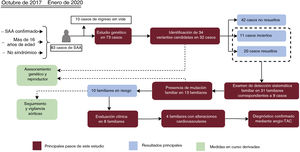
ResultadosEl análisis de los datos genómicos de 73 pacientes de SAA dio como resultado la identificación de 34 variantes candidatas en 32 individuos, localizadas en 14 genes diferentes. La segregación familiar se realizó en 31 individuos pertenecientes a 9 familias, donde se encontraron 13 portadores de los que 10 mostraron un genotipo compatible con SAA. El estudio estadístico indicó que la ausencia de hipertensión, una menor edad, una historia familiar de SAA y la ausencia de dolor están asociadas con un estudio genético positivo.
ConclusionesSe amplió el espectro mutacional asociado a SAA. Además, tanto los pacientes índice como los familiares estudiados se han visto beneficiados por estos resultados, por lo que se puede establecer el protocolo de seguimiento adecuado para cada uno de ellos. Por último, es importante destacar la posibilidad de utilizar variables clínicas estadísticamente significativas como factores predictores del carácter hereditario del SAA.
ClinicalTrials.gov (Identifier: NCT04751058)
Palabras clave
Los síndromes aórticos agudos (SAA) engloban 3 afecciones que ponen en peligro la vida, como son la disección aórtica aguda, el hematoma intramural y la úlcera aórtica penetrante1. En España, el SAA se da en entre 20 y 40 casos/millón de habitantes/año y se detecta con mayor frecuencia en los varones (73%)2. El 70% de estos casos afectan a la aorta ascendente, el 30% a la parte descendente, y en el 78,7% el mecanismo fundamental es la disección3. Solo alrededor de un 5% de los aneurismas de la aorta torácica son sintomáticos antes de que aparezca una complicación con peligro para la vida que surge de forma impredecible y se manifiesta por una muerte súbita cardíaca4. Así pues, un diagnóstico en el momento oportuno es esencial y permite salvar vidas1. Clásicamente, hay diversos factores de riesgo que se han asociado al SAA, como la hipertensión (76,6%), la enfermedad aterosclerótica (27%), el consumo de cocaína (1,8%) o el tabaquismo y la dislipidemia5. De igual modo, el SAA puede estar involucrado en cualquier trastorno asociado a alteraciones estructurales de la pared aórtica, como las conectivopatías, los aneurismas previos, la presencia de una válvula aórtica bicúspide6 o la lesión aórtica directa tras la cirugía o una intervención percutánea.
El SAA puede formar parte de síndromes genéticos bien caracterizados, como el síndrome de Marfan, el síndrome de Loeys-Dietz o el síndrome de Ehlers-Danlos vascular. Además, el SAA puede manifestarse en forma de una enfermedad aislada, limitada a la aorta torácica (SAA no sindrómico) y puede ser de carácter esporádico (80% de los SAA no sindrómicos4,7) o hereditario, dependiendo de la presencia de otros familiares afectados. El SAA es una enfermedad genéticamente heterogénea, con formas de presentación clínica solapadas; en consecuencia, los datos genéticos de los pacientes han pasado a ser cruciales durante el diagnóstico, para la identificación de otros familiares en riesgo8 y para el manejo personalizado del paciente, incluida la elección del momento oportuno para una reparación quirúrgica profiláctica9.
Tras la introducción de la secuenciación de nueva generación (NGS), se han alcanzado logros notables en el descubrimiento de variantes genéticas y de las moléculas reguladoras clave asociadas al SAA7,10–12. Hasta la fecha, se han asociado al SAA variantes situadas en alrededor de 40 genes, que suponen un 20% a 30% del total de casos. Se ha demostrado una transmisión autosómica dominante en la inmensa mayoría de los pacientes con la enfermedad, si bien se ha observado también una transmisión autosómica recesiva y una herencia ligada al cromosoma X13.
Las aortopatías tienen una mayor probabilidad de asociarse a una causa genética cuando la persona afectada tiene menos de 50 años o antecedentes familiares de aneurisma aórtico o disección aórtica o de muerte súbita inexplicada14. Además, la presencia de algunas manifestaciones sistémicas puede sugerir la presencia de una aortopatía torácica heredable sindrómica.
En este estudio analizamos la prevalencia de las variantes genéticas en un grupo de pacientes con SAA no sindrómico ingresados en la unidad de cuidados intensivos, con objeto de explorar la utilidad clínica de las variantes asociadas al SAA en la toma de decisiones terapéuticas y en la trazabilidad familiar de esta enfermedad clínicamente silente y causante de una gran morbilidad.
MÉTODOSCohorte de pacientes y vigilanciaSe llevó a cabo un estudio de observación prospectivo en un grupo de pacientes consecutivos ingresados en la unidad de cuidados intensivos del Hospital Universitario Virgen del Rocío de Sevilla, con un diagnóstico de SAA entre octubre de 2017 y enero de 2020. El estudio fue autorizado por el Portal de ética de la Investigación biomédica de Andalucía (PEIBA) (código interno del GEN-AOR 2436-N-20, ensayos clínicos [NCT04751058]). Se obtuvo un consentimiento informado para la inclusión en el estudio genético y para fines de investigación de todos los pacientes o sus tutores legales si procedía.
Las manifestaciones clínicas que se consideró que eran signos que sugerían un SAA durante la evaluación inicial de los pacientes fueron las siguientes: signos de dolor torácico intenso con una irradiación típica, hipertensión, pulso deficitario, diferencia de la lectura de la presión arterial entre los dos brazos, semiología de insuficiencia aórtica, signos de fallo ventricular, electrocardiograma inespecífico (normal o con hipertrofia ventricular izquierda), radiografía normal o con ensanchamiento del mediastino y otros signos clínicos que pudieran alertar al clínico respecto a la presencia de un síndrome de Marfan subyacente con el empleo de la escala de Gante. La confirmación del SAA se realizó con el empleo de una angiografía de tomografía computarizada (angio-TAC) multicorte (cortes de 1mm), con y sin el empleo de contraste yodado, para su posterior reconstrucción. Después de la confirmación y el análisis de las imágenes, se trató a los pacientes según las recomendaciones de la guía de la Sociedad Europea de Cardiología15 para el segmento afectado y para las características angiográficas halladas.
Tras la evaluación clínica y las exploraciones de imagen, se incluyó en el estudio genético a los pacientes que cumplían los siguientes criterios: a) SAA confirmado mediante angio-TAC; b) edad >16 años y c) ausencia de signos compatibles con trastornos sindrómicos. Se excluyó, en cambio, a los pacientes que ingresaron sin vida. Una vez seleccionada la cohorte de pacientes, se utilizó el registro de historias clínicas electrónicas para obtener información sobre sus antecedentes personales y familiares, la forma de presentación clínica y el curso de la enfermedad. A este respecto, los antecedentes familiares solamente se consideraron positivos si se identificaban otros familiares con aortopatías o muerte súbita.
Se recomendó un análisis de segregación familiar para todos los casos en los que se detectó una variante genética, incluidas las variantes probablemente patogénicas y las variantes de significado incierto. Después del asesoramiento genético, se evaluó mediante una ecocardiografía a los familiares que aceptaron una exploración clínica después de un resultado positivo o incierto en la prueba genética. Se utilizó angio-TAC durante la evaluación clínica únicamente después de observar una alteración en la ecocardiografía. Todos los familiares positivos fueron remitidos a los departamentos de asistencia integral de cardiopatías familiares y de enfermedades raras de nuestro centro.
Aislamiento de ADN y análisis de calidadSe realizó una extracción automática de ácido desoxirribonucleico (ADN) genómico con el empleo de un instrumento Chemagic 360 (Perkin Elmer, Estados Unidos). La integridad y la pureza del ADN se verificaron mediante electroforesis en gel y con el empleo de métodos tanto fluorimétricos como espectrofotométricos. Puede consultarse una descripción detallada del proceso completo en el apartado de métodos del material adicional.
Diseño del panel de genes de aortopatíasSe utilizó un panel de captura (Roche, Estados Unidos) que incluía 42 genes (tabla 1 del material adicional) relacionados con diferentes aortopatías. La selección de los genes capturados se basó en las guías actuales, las bases de datos públicas y el consenso de expertos. Puede consultarse una información más detallada al respecto en el apartado de métodos del material adicional.
Preparación de la biblioteca y generación de los datos de secuenciaciónSe utilizó un microgramo de ADN genómico para cada preparación de biblioteca utilizando el SeqCap EZ Library SR versión 5.1 (Roche, Estados Unidos) según el protocolo del fabricante, con modificaciones de carácter menor (apartado de métodos del material adicional). La secuenciación se llevó a cabo en la plataforma Illumina NextSeq500 (Illumina, Estados Unidos).
Análisis bioinformáticoEl análisis de los datos se realizó con el empleo de un protocolo validado con anterioridad16, con algunas modificaciones (apartado de métodos del material adicional).
Priorización de las variantes y evaluación de la patogenicidadLas variantes se priorizaron mediante criterios clínicos y genéticos establecidos (apartado de métodos del material adicional). Finalmente, solo se presentaron las variantes clasificadas como patogénicas, probablemente patogénicas o de significado incierto.
Análisis estadísticoLas variables continuas se expresan en forma de media±desviación estándar, mientras que las variables discretas se presentan mediante número absoluto y porcentaje. Todos los casos se clasificaron en los 2 grupos siguientes: a) individuos con variantes claramente patogénicas o probablemente patogénicas y b) individuos sin variantes o con variantes de significado clínico incierto. Las diferencias existentes entre los dos grupos en cuanto a las variables discretas se estimaron con la prueba exacta de Fisher, excepto en el caso de tablas de contingencia mayores de 2x2, para las que se utilizó la prueba exacta de Fisher-Freeman-Halton. Los resultados obtenidos se consideraron estadísticamente significativos para valores de p bilaterales <0,05. Todos los análisis estadísticos se llevaron a cabo con el empleo del programa IBM SPSS Statistics for Windows, versión 25.0 (IBM Corp., Estados Unidos).
Para evaluar la exactitud diagnóstica de las variantes de relevancia clínica y utilizar su combinación como herramienta de predicción de la heredabilidad, se calcularon los siguientes atributos17,18: tasa de positivos verdaderos (o sensibilidad), tasa de negativos verdaderos (o especificidad), tasa de falsos positivos, tasa de falsos negativos y odds ratio (OR), que representa la probabilidad esperada de que un caso con una variable clínica dada sea genéticamente positivo frente a la probabilidad de que un caso sin esa variable clínica tenga una prueba genética positiva. Un valor de OR superior a 1 implica una probabilidad de que un caso genético positivo manifieste una determinada variable superior a la probabilidad de que no la manifieste. Es de destacar que los casos clasificados como inciertos no se incluyeron en estos cálculos.
RESULTADOSCaracterísticas demográficas y clínicas de los casos de síndrome aórtico agudoEntre octubre de 2017 y enero de 2020, hubo un total de 83 pacientes con SAA ingresados en la unidad de cuidados intensivos que cumplieron los criterios de inclusión en el estudio genético. Sin embargo, 10 de ellos fallecieron en las primeras horas, sin posibilidad de obtener muestras de ADN, con lo que quedaron en total 73 pacientes para el estudio. En la tabla 1 se muestran las características clínicas generales de este grupo de pacientes, mientras que en la tabla 2 del material adicional se presenta una información detallada para cada uno de los casos. La mediana de edad fue de 57 años, con una relación varones/mujeres de aproximadamente 3:1. Con el empleo de la clasificación de Stanford, el 63% de los casos se encontraban en la clase Stanford A y el 37%, en la clase Stanford B. La disección (74%) fue la forma clínica más frecuente. La hipertensión y el tabaquismo fueron los factores de riesgo más comunes, y se observaron de manera aún más frecuente en los pacientes con una disección de aorta de tipo B (70,3% y 81,4%, respectivamente). En 57 pacientes, el primer síntoma fue el dolor, acompañado de una crisis hipertensiva en 27 de ellos. En 60 pacientes (82,2%) se utilizó un tratamiento quirúrgico, con carácter de emergencia o urgencia en el 46,2% de los casos. Es de destacar que el 47,9% de los pacientes tenían un diámetro aórtico inferior a 5cm.
Características del grupo del estudio
| Número/media±DE | Porcentaje/intervalos | p | |
|---|---|---|---|
| Descripción de los pacientes | |||
| Total de pacientes | 73 | 100 | - |
| Edad de presentación del SAA a | 57±13,16 | 16-81 | - |
| <55 años | 33 | 45,2 | < 0,001c |
| Varones | 54 | 74 | 0,241 |
| Mujeres | 19 | 26 | |
| Geografía, raza | |||
| Andalucía | 70 | 95,8 | 0,557 |
| Otras | 3 | 4,2 | |
| Raza caucásica | 73 | 100 | - |
| Tipo de presentación clínica | |||
| Aguda (<15 d) | 54 | 74 | 1,000 b |
| Subaguda (> 15 d) | 7 | 9,6 | |
| Crónica (> 1 mes) | 12 | 16,4 | |
| Stanford A | 46 | 63 | 0,278 |
| Stanford B | 27 | 37 | |
| Diámetro de la aorta, cma | 4,57±1,5 | 2,5-12,5 | - |
| Tipo | |||
| Disección | 52 | 74 | 0,562 b |
| Úlcera penetrante | 5 | 6,8 | |
| Hematoma intramural | 7 | 9,6 | |
| Ruptura de aneurisma | 2 | 2,7 | |
| Aneurisma | 7 | 9,6 | |
| Factores de riesgo | |||
| Tabaquismo | 44 | 60,3 | 0,424 |
| Hipertensión | 42 | 57,3 | 0,007 c |
| Cirugía cardiaca | 3 | 4,1 | 0,557 |
| DM | 5 | 6,8 | 0,610 |
| EPOC | 8 | 11 | 0,432 |
| Antecedentes familiares | 15 | 20,5 | 0,003 c |
| Hiperlipidemia | 19 | 26 | 0,241 |
| Abuso de drogas | 15 | 20,5 | 0,267 |
| Válvula bicúspide | 2 | 2,7 | 1,000 |
| Síntomas clínicos | |||
| Dolor | 57 | 78,1 | 0,009 c |
| Crisis hipertensiva | 28 | 38,4 | 1,000 |
| Pulso deficitario | 23 | 31,5 | 1,000 |
| Shock | 13 | 17,8 | 0,326 |
| Insuficiencia renal aguda | 21 | 28,8 | 0,393 |
| Taponamiento cardiaco | 6 | 8,2 | 0,663 |
| Isquemia de órganos internos | 12 | 16,4 | 1,000 |
| Síncope | 14 | 19,4 | 0,744 |
| Tratamiento | |||
| Tratamiento médico | 13 | 17,8 | 0,742 |
| Tratamiento quirúrgico | 60 | 82,2 | |
| Cirugía abierta | 40 | 66,6 | 0,371 |
| Endovascular | 20 | 27,4 | |
| Criterios de tratamiento | |||
| De emergencia | 28 | 38,4 | 0,833 b |
| Urgente | 13 | 17,8 | |
| Demorado | 19 | 26 | |
| Estancia en la UCIa | 11±11,2 | 1-51 | - |
| Estancia en el hospitala | 21±17,5 | 1-70 | - |
| Muertes relacionadas con SAA | 14 | 19,2 | 0,747 |
SAA: síndrome aórtico agudo; DM: diabetes mellitus; EPOC: enfermedad pulmonar obstructiva crónica; UCI: unidad de cuidados intensivos; DE: desviación estándar.
Los datos se expresan en forma de media±desviación estándar e intervalos de confianza para las variables continuas (a) y de número absoluto y porcentaje para las variables discretas. Los valores de p indican la significación obtenida aplicando la prueba estadística (prueba exacta de Fisher o prueba exacta de Fisher-Freeman-Halton (b), según proceda) para relacionar los resultados genéticos con las variables discretas, y las marcadas con una letra c son estadísticamente significativas (c).
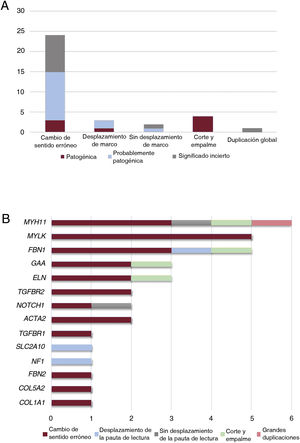
Nuestra aplicación de NGS nos permitió abarcar el 97,1% de las bases diana, con lo que hubo una cobertura media de 357,9x y un porcentaje de lecturas mapeadas en la diana del 83,4%. Todas las regiones codificantes correspondientes a genes asociados a aortopatías (tabla 1 del material adicional) se cubrieron en > 20x. El análisis de los datos de NGS condujo a la detección de 34 variantes genéticas candidatas heterocigotas en un total de 32 pacientes (tabla 2 y figura 1), incluidas 24 de cambio de sentido, 4 de corte y empalme, 3 que provocan un desplazamiento de la pauta de lectura, 2 deleciones que no afectan al desplazamiento de la pauta de lectura y 1 variación en el número de copias (figura 2A).
Mutaciones causantes probables y variantes de significado incierto identificadas en la cohorte del estudio
| ID | Gen | Mutación | Presencia en GnomAD | Ref. | Relevancia clínica notificada | Herramientas de predicción in silico | ACMG | Nuestra clasificación | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cDNA | Proteína | Sift | PPhen | CADD | HSF | MutTast | |||||||
| 3 | MYH11 | c.3766_3768del | p.Lys1263del | 13 het | 1 | PP a,b | - | - | - | - | CE | VSI | PP |
| 8 | MYLK | c.3929T> G | p.Leu1310Arg | Ninguna | Nueva | n. d. | D | PD | P | - | CE | VSI | PP |
| 10 | GAA | c.-32-13T> G | n. d. | 856 het/1 hom | 2 | P a,b | - | - | B | - | CE | P | P |
| c.2237G> C | p.Trp746Ser | 20 het | 3 | P a,b | D | PD | P | - | CE | P | P | ||
| 12 | MYLK | c.4749C> A | p.His1583Gln | Ninguna | Nueva | n. d. | D | PsD | P | - | CE | VSI | PP |
| 15 | FBN1 | c.7334T> C | p.Leu2445Pro | Ninguna | Nueva | n. d. | D | PD | P | - | CE | PP | PP |
| 18 | FBN1 | c.7412C> G | p.Pro2471Arg | 7 het | 4 | VSI a,b,c | T | PD | P | - | CE | VSI | VSI |
| 19 | MYH11e | Duplicación de 16p13.11 | n. d. | n. d. | 5 | IPC b,d | - | - | - | - | - | VSI | VSI |
| 22 | MYLK | c.5329C> T | p.Pro1777Ser | 4 het | rs1 | VSI a | D | PD | P | - | CE | VSI | VSI |
| 27 | ELN | c.1150+1G> A | n. d. | 17 het | 7 | P a | - | - | P | WSB | CE | P | P |
| 28 | COL1A1 | c.2995C> T | p.Pro999Ser | Ninguna | Nueva | n. d. | T | PD | P | - | CE | PP | PP |
| 29 | NOTCH1 | c.4372G> A | p.Ala1458Thr | 23 het | rs2 | VSI a | T | B | B | - | PM | B | VSI |
| 31 | MYLK | c.2149G> A | p.Asp717Asn | 25 het | 8 | VSI a,h | D | PD | P | - | CE | VSI | VSI |
| 35 | ACTA2 | c.897T> G | p.Asn299Lys | Ninguna | Nueva | n. d. | D | PsD | P | - | CE | VSI | PP |
| 38 | MYH11 | c.2156G> A | p.Arg719Gln | Ninguna | 9 | P a,b | D | PD | P | - | CE | PP | P |
| 39 | MYLK | c.1315G> A | p.Gly439Arg | 7 het | rs3 | VSI a | D | PsD | P | - | CE | VSI | PP |
| 40 | TGFBR2 | c.1061C> T | p.Ala354Val | 7 het | rs4 | n. d. h | T | B | P | - | CE | VSI | VSI |
| 44 | ACTA2 | c.412A> G | p.Ile138Val | Ninguna | Nueva | n. d. | T | PD | P | - | CE | VSI | PP |
| 48 | NOTCH1 | c.2263_2265del | p.Asn755del | 60 het | rs5 | VSI a,c | - | - | - | - | CE | B | VSI |
| 49 f | GAA | c.2237G> C | p.Trp746Ser | 20 het | 10 | P a,b,c | D | PD | P | - | CE | P | P |
| 52 | ELN | c.1921C> A | p.Leu641Ile | Ninguna | Nueva | n. d. | T | PD | B | - | PM | PB | VSI |
| 53 | SLC2A10 | c.1334del | p.Gly445Glufs*40 | 24 het | 11 | P a,b,c | - | - | - | - | CE | P | P |
| 60 | ELN | c.1288G> A | p.Gly430Arg | 6 het | rs6 | n. d. | T | PD | B | - | PM | PB | VSI |
| 61 | MYH11 | c.4599+2dup | n. d. | Ninguna | rs7 | P a | - | - | - | WSB | CE | VSI | P |
| 62 | FBN2 | c.2671A> G | p.Ile891Val | Ninguna | Nueva | n. d. | T | B | B | - | CE | VSI | VSI |
| 63 | FBN1 | c.8517del | p.Glu2841Asnfs*4 | Ninguna | Nueva | n. d. | - | - | - | - | CE | P | PP |
| 6g | NF1 | c.2816del | p.Asn939Ilefs*14 | Ninguna | 12 | P b | - | - | - | - | CE | P | PP |
| 65 | COL5A2 | c.2806G> A | p.Gly936Arg | Ninguna | rs8 | n. d. | D | PD | P | - | CE | VSI | PP |
| 67 | FBN1 | c.5917+1G> T | n. d. | Ninguna | rs9 | P a,c | - | - | P | WSB | CE | P | P |
| 68 | MYH11 | c.3397G> C | p.Ala1133Pro | Ninguna | Nueva | n. d. | D | PD | P | - | CE | VSI | PP |
| 69 | FBN1 | c.1130G> T | p.Cys377Phe | Ninguna | Nueva | n. d. h | D | PD | P | - | CE | PP | PP |
| 71 | MYH11 | c.214G> A | p.Gly72Arg | Ninguna | Nueva | n. d. | T | B | P | - | CE | PB | VSI |
| 72 | TGFBR1 | c.934G> T | p.Gly312Cys | Ninguna | Nueva | n. d. h | D | PD | P | - | CE | PP | PP |
| TGFBR2 | c.1067G> A | p.Arg356Gln | 5 het | rs10 | VSI a,c | T | B | P | - | CE | PP | PP | |
ACMG: clasificación de las variantes basada en los criterios del American College of Medical Genetics and Genomics; B: benigna; CADD: combined annotation dependent depletion (se consideran patogénicas las variantes con una puntuación ≥ 15); IPC: interpretación de patogenicidad contradictoria; D: deletérea; CE: causante de enfermedad; Het: heterocigotos; Hom: homocigotos; HSF: human splicing finder; ID: número de identificación; PB: probablemente benigna; PP: probablemente patogénica; MutTast: mutation taster; n. d.: no disponible; Nuestra clasificación: clasificación de variantes basada en toda la información obtenida; P: patogénica; PD: probablemente deletérea; PM: polimorfismo; Pphen: Pphen-2; PsD: posiblemente deletérea; Ref: referencias; rs1, rs748200926; rs2, rs200495793; rs3, rs190877071; rs4, rs376752333; rs5, rs587778559; rs6, rs274470120; rs7, rs794728677; rs8, rs1471655044; rs9, rs363808; rs10, rs727504292; T: tolerada; VSI: variante de significado incierto; WSB: rotura en la posición silvestre de corte y empalme.
Todas las variantes detectadas se hallaron en estado heterocigoto. La relevancia clínica notificada se verificó tanto en las bases de datos ClinVar (a), LOVD (b) y HGMD (c) como en la literatura (d).
Variación del número de copias que afecta a la región genómica incluido el gen MYH11 y loci adicionales.
Caso no resuelto debido a que solo se identificó 1 variante heterocigota en un gen autosómico recesivo.

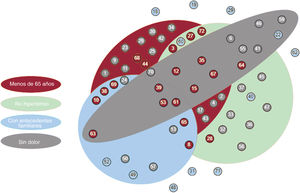
Figura central. Diagrama de flujo de los pacientes, las pruebas de detección sistemática de los familiares y los resultados de las pruebas genéticas. Los recuadros de color naranja indican los pasos principales llevados a cabo en nuestro proceso de trabajo, mientras que los resultados principales se indican en recuadros de color rojo. Las medidas en curso aplicadas como consecuencia de este estudio se indican en recuadros de color verde. SAA, síndrome aórtico agudo; Angio-TAC, angiografía de tomografía computarizada multicorte.

Clasificación de las 34 variantes identificadas asociadas a una aortopatía en nuestra cohorte. A: tipo de variantes según el tipo de consecuencia. B: distribución de los tipos de variantes, incluidas las de cambio de sentido, desplazamiento de la pauta de lectura, ausencia de desplazamiento de la pauta de lectura, corte y empalme, y grandes duplicaciones en los genes asociados a la aortopatía en nuestra cohorte.
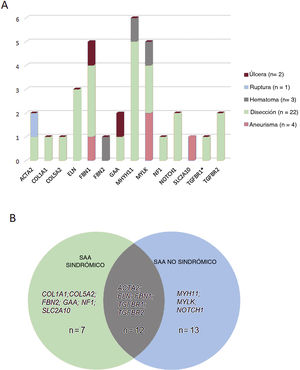
Estas variantes se hallaron en 14 genes diferentes, siendo el gen MYH11 el que mostró una mayor prevalencia de mutación, seguido del MYLK y del FBN1 (figura 2B). Aunque estos genes intervienen en un amplio espectro de vías moleculares (tabla 1 del material adicional), la mayoría de ellos (22 casos, 69%) se asociaron a una disección aórtica en nuestra cohorte (figura 3A). En todos los casos, nuestros resultados fueron compatibles con un patrón de transmisión hereditaria autosómica dominante excepto para la familia 10, en la que la heterocigosidad compuesta con 2 cambios en el GAA se consideró la causa más probable de la enfermedad.

Correlaciones entre genotipo y fenotipo. A: esquema del tipo de aortopatía asociada a los genes identificados en este estudio. B: número de casos portadores de variantes en genes asociados a formas de aortopatía sindrómicas, no sindrómicas o de ambos tipos. El asterisco indica una variante en el gen TGFBR1 presente en 1 de los casos portadores de una variante concomitante en el gen TGFBR2.
Aproximadamente el 40,7% de los pacientes (n=13) tenían variantes genéticas en genes que se han asociado previamente a formas familiares no sindrómicas, un 21,8% (n=7) a trastornos sindrómicos y un 37,5% (n=12) eran portadores de variantes en genes asociados a formas tanto sindrómicas como no sindrómicas (figura 3B). Tiene interés señalar que 14 casos a priori no sindrómicos albergaban variantes patogénicas o probablemente patogénicas en genes que se han asociado previamente a una posible enfermedad aórtica sindrómica. Por consiguiente, a los pacientes 10, 53, 64 y 69 se les diagnosticó una enfermedad de Pompe, síndrome de tortuosidad arterial, neurofibromatosis y síndrome de Marfan, respectivamente, y a los pacientes 28 y 65 se les diagnosticó un síndrome de Ehler-Danlos. Esto nos permitió reclasificar el 18,7% de los casos como aortopatías sindrómicas, lo cual tiene consecuencias importantes para el manejo tanto del paciente como de los familiares.
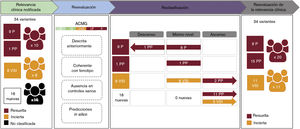
Evaluación de la relevancia clínica de las variantes identificadasDiez de las 34 variantes identificadas se habían descrito anteriormente como patogénicas o probablemente patogénicas en bases de datos públicas o en la literatura, mientras que 8 habían sido descritas anteriormente como variantes de significado incierto, y 16 eran variantes nuevas sin ninguna asociación clínica hasta ese momento (tabla 2 y figura 4). Se llevó a cabo una reevaluación de las variantes según las asociaciones clínicas previas, el recuento de alelos en gnomAD, las predicciones de patogenicidad in silico y los criterios del American College of Medical Genetics and Genomics, con lo que se llegó a la reclasificación de 19 variantes (figura 4). Esto condujo a la identificación de 11 variantes de significado incierto en 11 casos (15,07%) y de 23 variantes patogénicas o probablemente patogénicas en 21 pacientes, aunque tan solo 20 casos se consideraron resueltos (27,39%), ya que se halló una única variante heterocigota en el gen GAA autosómico recesivo en el paciente 49. Si se confirma una asociación clínica para estas variantes, la tasa diagnóstica de este estudio podría alcanzar el 42,46%.

Proceso de trabajo para la reevaluación, clasificación e interpretación clínica de la variante. ACMG: clasificación de las variantes basada en los criterios del American College of Medical Genetics and Genomics; B: benigna; PB: probablemente benigna; PP: probablemente patogénica; P: patogénica; VSI: variante de significado incierto.
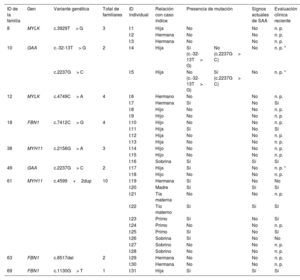
Hubo familiares pertenecientes a 9 familias en las que se detectaron variantes candidatas que aceptaron ser estudiados (tabla 3 y figura 1). Confirmamos la presencia de las variantes detectadas en 13 de los 31 familiares analizados mediante secuenciación Sanger. Tres (I:4, I:5 e I:17) de los 13 familiares eran portadores de variantes genéticas asociadas a un rasgo autosómico recesivo y, por consiguiente, sus genotipos no eran compatibles con un fenotipo de SAA. Los 10 familiares restantes eran portadores de variantes genéticas compatibles con una patrón de herencia autosómica dominante, por lo que sus genotipos eran coherentes con la presencia de un SAA.
Segregación familiar de las variantes genéticas identificadas en los 9 pacientes en los que se dispuso de familiares
| ID de la familia | Gen | Variante genética | Total de familiares | ID individual | Relación con caso índice | Presencia de mutación | Signos actuales de SAA | Evaluación clínica reciente | |
|---|---|---|---|---|---|---|---|---|---|
| 8 | MYLK | c.3929T> G | 3 | I:1 | Hija | No | No | n. p. | |
| I:2 | Hermana | No | No | n. p. | |||||
| I:3 | Hermana | No | No | n. p. | |||||
| 10 | GAA | c.-32-13T> G | 2 | I:4 | Hija | Sí (c.-32-13T> G) | No (c.2237G> C) | No | n. p. * |
| c.2237G> C | I:5 | Hija | No (c.-32-13T> G) | Sí (c.2237G> C) | No | n. p. * | |||
| 12 | MYLK | c.4749C> A | 4 | I:6 | Hermano | No | No | n. p. | |
| I:7 | Hermana | Sí | No | Sí | |||||
| I:8 | Hijo | No | No | n. p. | |||||
| I:9 | Hijo | No | No | n. p. | |||||
| 18 | FBN1 | c.7412C> G | 4 | I:10 | Hijo | No | No | n. p. | |
| I:11 | Hija | Sí | No | Sí | |||||
| I:12 | Hija | No | No | n. p. | |||||
| I:13 | Hija | No | No | n. p. | |||||
| 38 | MYH11 | c.2156G> A | 3 | I:14 | Hijo | No | No | n. p. | |
| I:15 | Hijo | No | No | n. p. | |||||
| I:16 | Sobrina | Sí | Sí | Sí | |||||
| 49 | GAA | c.2237G> C | 2 | I:17 | Hija | Sí | No | n. p. * | |
| I:18 | Hijo | No | No | n. p. | |||||
| 61 | MYH11 | c.4599+2dup | 10 | I:19 | Hermana | Sí | No | No | |
| I:20 | Madre | Sí | Sí | Sí | |||||
| I:21 | Tía materna | No | No | n. p. | |||||
| I:22 | Tío materno | Sí | Sí | Sí | |||||
| I:23 | Primo | Sí | No | Sí | |||||
| I:24 | Primo | No | No | n. p. | |||||
| I:25 | Primo | Sí | No | Sí | |||||
| I:26 | Sobrina | Sí | No | No | |||||
| I:27 | Sobrino | No | No | n. p. | |||||
| I:28 | Sobrino | No | No | n. p. | |||||
| 63 | FBN1 | c.8517del | 2 | I:29 | Hermana | No | No | n. p. | |
| I:30 | Hermana | No | No | n. p. | |||||
| 69 | FBN1 | c.1130G> T | 1 | I:31 | Hija | Sí | Sí | Sí | |
SAA: síndrome aórtico agudo; ID: número de identificación; n. p.: no procede.
Todos los individuos portadores de mutaciones son heterocigotos. Los individuos con signos actuales de SAA mostraron alteraciones cardiovasculares en las exploraciones de imagen más recientes. Se ofreció una evaluación clínica reciente a los pacientes en los que se detectó un genotipo causal, sin que ello fuera aplicable a los pacientes con el tipo natural ni a los portadores de variantes autosómicas recesivas (*).
Se ofreció una nueva evaluación clínica a los 10 familiares con un resultado positivo, pero solamente 8 de ellos acudieron a la cita. Cuatro de estos últimos presentaban ya alteraciones cardiovasculares: a) el individuo I:16 (16 años) presentaba una actividad eléctrica cardiaca en reposo con un QTc en el límite superior (464ms); b) el individuo I:20 (67 años) presentaba una dilatación leve de la raíz aórtica (37mm) y de la aorta ascendente (40mm) y una insuficiencia aórtica de grado II; c) el individuo I:21 (63 años) presentaba una dilatación leve de la raíz aórtica (43mm) y de la aorta ascendente (38mm), y una ateromatosis calcificada difusa leve; y d) el individuo I:31 (15 años) presentaba una dilatación leve de la arteria pulmonar. Es de destacar que, con la excepción del individuo I:7, todos los familiares con resultados genéticos positivos y sin signos actuales de aortopatías eran más jóvenes que el paciente índice de su familia. De hecho, 11 de los 13 familiares en los que se detectó una variante de tipo familiar se encuentran actualmente en edad reproductiva (< 50 años).
Correlación de los resultados genéticos con las variables clínicasLas pruebas estadísticas mostraron la existencia de diferencias significativas entre los pacientes con un diagnóstico genético claro y los pacientes sin variantes causales o inciertas, para las siguientes variables clínicas: hipertensión, edad del paciente, antecedentes familiares y dolor (tabla 1 y figura 5). La ausencia de hipertensión, una edad <55 años, la presencia de antecedentes familiares de SAA y sin dolor como primer síntoma mostraron una correlación significativa con la detección de variantes genéticas patogénicas o probablemente patogénicas y, por consiguiente, con casos hereditarios.
, variantes de significado incierto (azul) o no se identificaron variantes candidatas (gris).")
Asociación entre los casos de síndrome aórtico agudo hereditario y las manifestaciones clínicas significativas. Cada círculo corresponde a 1 de las familias en estudio, y se indica su número de identificación. El color de los círculos indica si se hallaron variantes patogénicas o probablemente patogénicas (rojo), variantes de significado incierto (azul) o no se identificaron variantes candidatas (gris).
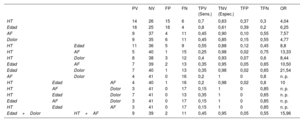
Todos los casos resueltos se incluyeron en al menos 1 de estos 4 grupos estadísticamente significativos, mientras que la combinación de las 4 variables significativas englobó tan solo 3 de estos casos (figura 5). De hecho, en la tabla 4 se muestra que, mientras que la sensibilidad fue máxima para la edad y la hipertensión (una tasa de positivos verdaderos del 70% y 80%, respectivamente), los antecedentes familiares y el dolor fueron los factores con mayor especificidad (una tasa de negativos verdaderos del 90% y 85%, respectivamente). La combinación de estas variables clínicas nos permitió obtener un buen equilibrio entre sensibilidad y especificidad, así como mejorar su capacidad predictiva en función del cálculo de la OR. Si analizamos los valores máximos de este atributo, la combinación de edad y dolor (OR=21,54) y la combinación de hipertensión y antecedentes familiares (OR=13,33) serían los conjuntos con una mayor fuerza predictiva, aunque su sensibilidad se reduciría (35% y 25%, respectivamente). Sin embargo, el empleo conjunto de estas 2 combinaciones nos permitió mantener un valor predictivo alto (OR=15,96) sin reducir excesivamente la sensibilidad (45%) y garantizando la especificidad (95%).
Atributos calculados para medir la exactitud diagnóstica de las variables clínicas estadísticamente significativas
| PV | NV | FP | FN | TPV (Sens.) | TNV (Espec.) | TFP | TFN | OR | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HT | 14 | 26 | 15 | 6 | 0,7 | 0,63 | 0,37 | 0,3 | 4,04 | |||
| Edad | 16 | 25 | 16 | 4 | 0,8 | 0,61 | 0,39 | 0,2 | 6,25 | |||
| AF | 9 | 37 | 4 | 11 | 0,45 | 0,90 | 0,10 | 0,55 | 7,57 | |||
| Dolor | 9 | 35 | 6 | 11 | 0,45 | 0,85 | 0,15 | 0,55 | 4,77 | |||
| HT | Edad | 11 | 36 | 5 | 9 | 0,55 | 0,88 | 0,12 | 0,45 | 8,8 | ||
| HT | AF | 5 | 40 | 1 | 15 | 0,25 | 0,98 | 0,02 | 0,75 | 13,33 | ||
| HT | Dolor | 8 | 38 | 3 | 12 | 0,4 | 0,93 | 0,07 | 0,6 | 8,44 | ||
| Edad | AF | 7 | 39 | 2 | 13 | 0,35 | 0,95 | 0,05 | 0,65 | 10,50 | ||
| Edad | Dolor | 7 | 40 | 1 | 13 | 0,35 | 0,98 | 0,02 | 0,65 | 21,54 | ||
| AF | Dolor | 4 | 41 | 0 | 16 | 0,2 | 1 | 0 | 0,8 | n. p. | ||
| HT | Edad | AF | 4 | 40 | 1 | 16 | 0,2 | 0,98 | 0,02 | 0,8 | 10 | |
| HT | AF | Dolor | 3 | 41 | 0 | 17 | 0,15 | 1 | 0 | 0,85 | n. p. | |
| HT | Edad | Dolor | 7 | 41 | 0 | 13 | 0,35 | 1 | 0 | 0,65 | n. p. | |
| Edad | AF | Dolor | 3 | 41 | 0 | 17 | 0,15 | 1 | 0 | 0,85 | n. p. | |
| HT | Edad | AF | 3 | 41 | 0 | 17 | 0,15 | 1 | 0 | 0,85 | n. p. | |
| Edad+Dolor | HT+AF | 9 | 39 | 2 | 11 | 0,45 | 0,95 | 0,05 | 0,55 | 15,96 |
AF: antecedentes familiares; FN: falsos negativos; TFN: tasa de falsos negativos; FP: falsos positivos; TFP: tasa de falsos positivos; HT: hipertensión; OR: odds ratio; NV: negativos verdaderos; TNV: tasa de negativos verdaderos o especificidad (Espec.); PV: positivos verdaderos; TPV: tasa de positivos verdaderos o sensibilidad (Sens.).
El tamaño muestral efectivo fue de 61, puesto que los casos con variantes de significado incierto no se incluyeron en los cálculos.
La aortopatía constituye un importante reto diagnóstico debido a su carácter silente; sin embargo, sus complicaciones constituyen una causa importante de gravedad extrema y de mortalidad. Dado que más del 20% de los pacientes refieren la presencia de unos antecedentes familiares positivos, la genética del SAA se ha investigado ampliamente como posible vía para el diagnóstico y para la estratificación del riesgo.
En este estudio utilizamos pruebas multigénicas basadas en NGS como método de detección sistemática para identificar variantes genéticas relacionadas con la enfermedad en el contexto del diagnóstico molecular de las aortopatías. La observación de diferencias notables en el rendimiento diagnóstico (del 3% al 37%) es frecuente en este grupo de enfermedades10,11,19, y se ha atribuido a diferencias en los criterios de inclusión de los pacientes, en las reglas de clasificación de las variantes y en la selección de los genes a incluir en el estudio. De manera coherente con los datos publicados con anterioridad 10,11,19, en el presente estudio alcanzamos una tasa diagnóstica del 27,39%, que puede aumentar al 42,46% si las variantes clasificadas como de significado incierto pasan a ser finalmente variantes probablemente patogénicas.
El análisis bioinformático nos permitió detectar 34 variantes genéticas candidatas heterocigotas en 14 genes asociados al SAA. El gen MYH11 fue el que mostró una mayor prevalencia de mutación, seguido de los genes MYLK y FBN1, lo cual es coherente con los datos de prevalencia publicados con anterioridad19,20. En cambio, las variantes genéticas en el gen ACTA2 están infrarrepresentadas (6,2%) en nuestra población en comparación con lo indicado por estudios previos (12%-21%)21, lo cual podría indicar un papel menos importante de este gen en nuestra población o un sesgo motivado por las diferencias en los criterios de inclusión.
El establecimiento de correlaciones entre genotipo y fenotipo se ve especialmente dificultado en estos trastornos a causa del amplio espectro de manifestaciones físicas, lo cual sugiere la intervención de factores de riesgo genéticos o ambientales multifactoriales10,22. Nuestros resultados respaldan el concepto de que un abordaje sin hipótesis previas y con independencia de supuestos clínicos, en genes cuidadosamente seleccionados por expertos, mejora considerablemente el manejo genético de estos pacientes23. Además, la mayor parte de las enfermedades autosómicas dominantes que suponen un riesgo para la vida de los pacientes presentan importantes dificultades en su diagnóstico antes de que se produzcan complicaciones4. En consecuencia, una limitación frecuente es el tamaño muestral y la disponibilidad de familiares afectados para poder realizar estudios de segregación concluyentes. De hecho, en este estudio, la segregación familiar solamente se llevó a cabo en 9 de las 13 familias, lo cual resalta la importancia de realizar reevaluaciones periódicas para afinar la clasificación de las variantes. Esta ardua tarea es de especial importancia cuando se detectan defectos genéticos concomitantes24. Es de destacar que se hallaron 2 variantes probablemente patogénicas en los genes TGFBR1 y TGFBR2, respectivamente, en el paciente 72; sin embargo, serán necesarios nuevos estudios para determinar el grado de intervención de cada uno de ellos en la aparición o la gravedad de la enfermedad.
Además, más allá de esclarecer las correlaciones entre genotipo y fenotipo y alcanzar un diagnóstico genético de los pacientes índice, las pruebas genéticas en el SAA desempeñan un papel crucial en la determinación del riesgo familiar25. Los familiares en riesgo asintomáticos, de mayor o menor edad de un individuo afectado deben ser evaluados con prontitud para esclarecer su estado genético y para establecer el grado apropiado de vigilancia. El rastreo genético para la aortopatía familiar en 31 familiares identificó un genotipo compatible con la aparición de un SAA en 10 individuos que están siendo objeto de seguimiento y tratamiento al aparecer los síntomas (I:16, I:20, I:21 e I:31). Además, otros 3 individuos (I:4, I:5 e I:17) eran portadores de variantes que es posible que causen un SAA en el contexto de un trastorno autosómico recesivo. Así pues, un total de 13 individuos pueden obtener también un beneficio con el asesoramiento genético reproductivo y las pruebas genéticas previas a la implantación, lo cual tiene especial importancia teniendo en cuenta que la mayor parte de los familiares estudiados están en edad reproductora. Además, la identificación de los familiares en riesgo evitó la vigilancia de 15 individuos en los que no había la variante causante de la enfermedad, con lo que se redujeron los costes asociados y se alivió la preocupación de esos pacientes. No obstante, la ausencia de una variante genética de significado clínico incierto en un individuo (familia 18) no es suficiente para no aplicar la vigilancia en ese individuo20.
Es importante señalar que el establecimiento de correlaciones entre genotipo y fenotipo no solo nos permite prever la aparición de los síntomas en los pacientes portadores de variantes genéticas causales, sino también predecir el carácter hereditario del SAA, a pesar de que no se haya identificado todavía el defecto genético subyacente. En este estudio, 4 características clínicas mostraron una asociación significativa con la presencia de variantes patogénicas. Mientras que variables clínicas como la presión arterial normal, la forma de presentación temprana de la enfermedad y los antecedentes familiares positivos se han asociado con anterioridad al SAA hereditario24,26, este estudio apunta también a la ausencia de dolor como indicador de una enfermedad de la aorta torácica heredable. De hecho, nosotros sugerimos que la combinación de las variables clínicas de edad y dolor, junto con la combinación de la hipertensión y los antecedentes familiares, puede aplicarse como herramienta predictiva útil para detectar la heredabilidad. Es de destacar que otras variables clínicas comúnmente asociadas a los resultados genéticos, como el tipo de disección aórtica27, no mostraron una asociación en este estudio, lo cual sugiere la existencia de diferencias entre las poblaciones o de un sesgo debido a nuestro tamaño muestral limitado.
CONCLUSIONESEn resumen, la aplicación de esta estrategia de NGS sirvió de guía para el manejo clínico de nuestros pacientes con aortopatías, al prevenir eventos causantes de emergencias médicas en los propios pacientes y en sus familiares y proporcionar un asesoramiento genético y reproductivo apropiado. A medida que las pruebas genéticas pasen a ser de uso más generalizado y tengan mayor exactitud, podría desarrollarse una biblioteca integral de variantes genéticas patogénicas, con lo que mejoraría la estratificación del riesgo, la posibilidad de intervenciones quirúrgicas personalizadas gracias a la información genética, la diferenciación entre el SAA y otros trastornos graves, y el tratamiento de los casos de SAA que plantean un reto clínico.
FINANCIACIÓNEste trabajo contó con el apoyo del Instituto de Salud Carlos III (ISCIII), Ministerio de Economía y Competitividad de España y fue cofinanciado por la Unión Europea (FEDER, «Una manera de hacer Europa ») [PI18-00612; PI19/01550; PI21-00244; IMP/00009], Consejería Regional de Salud y Familias del Gobierno Autónomo de Andalucía [PEER-0501-2019; PEER-0470-2019], Consejería Regional de Transformación Económica, Industria, Conocimiento y Universidades de Andalucía [P20_00887] y la Fundación Isabel Gemio/Fundación Cajasol [FGEMIO-2019-01]. NBG contó con el apoyo de la beca HR-0118-2020, financiada por la Consejería Regional de Salud y Familias del Gobierno Autónomo de Andalucía.
CONTRIBUCIÓN DE LOS AUTORESA.M. Puppo Moreno y S. Borrego concibieron el estudio. A.M. Puppo Moreno, S. Borrego, N. Bravo-Gil y C. Méndez-Vidal diseñaron el estudio y redactaron la versión inicial del manuscrito. A.M. Puppo Moreno, A. Adsuar Gómez, F. Tadeo Gómez Ruiz, C. Jiménez de Juan, R. Martín Bermúdez, J.M. López Sánchez, S. Martín Sastre y M. Fernández Caro obtuvieron los datos clínicos y se encargaron del tratamiento clínico. A.M. Puppo Moreno, P. Gallego y S. Borrego interpretaron los datos clínicos. N. Bravo-Gil, C. Méndez-Vidal y R.M. Fernández García analizaron e interpretaron los datos genómicos. S. Borrego, N. Bravo-Gil y C. Méndez-Vidal obtuvieron los datos genómicos. A. Adsuar Gómez, F. Tadeo Gómez Ruiz, C. Jiménez de Juan, R.M. Fernández García, R. Martín Bermúdez, J.M. López Sánchez, S. Martín Sastre, M. Fernández Caro y P. Gallego realizaron una revisión crítica del manuscrito respecto a contenido intelectual importante. Todos los autores aprobaron la versión final a publicar. A.M. Puppo Moreno, N. Bravo-Gil y C. Méndez-Vidal deben considerarse conjuntamente primeros autores.
CONFLICTO DE INTERESESTodos los autores expresan no tener nada que declarar.
- –
Las pruebas genéticas han pasado a ser un elemento crucial durante el diagnóstico, el tratamiento, la vigilancia, la estratificación del riesgo y la evaluación familiar de los pacientes con SAA.
- –
Hasta la fecha hay variantes en alrededor de 40 genes que se han asociado al SAA.
- –
Continúan surgiendo correlaciones específicas entre genotipo y fenotipo, lo cual permite un manejo y un tratamiento más precisos e individualizados de los pacientes con SAA.
- –
La identificación de nuevas variantes asociadas al SAA, con lo que se amplía nuestro conocimiento de la base genética de este trastorno.
- –
La descripción de la forma en la que los resultados genéticos pueden contribuir a elaborar las estrategias destinadas a prevenir y tratar el SAA.
- –
La posibilidad de predecir el carácter heredable del SAA a través de la evaluación y la combinación de características clínicas como la hipertensión, la edad, el dolor y los antecedentes familiares.